Lemey Philippe, Suchard Marc, Rambaut Andrew
UCLA and Institute of Evolutionary Biology, University of Edinburgh, Edinburgh & The Fogarty International Center, NIH.
PLoS Curr. 2009 Sep 2;1:RRN1031. doi: 10.1371/currents.RRN1031.
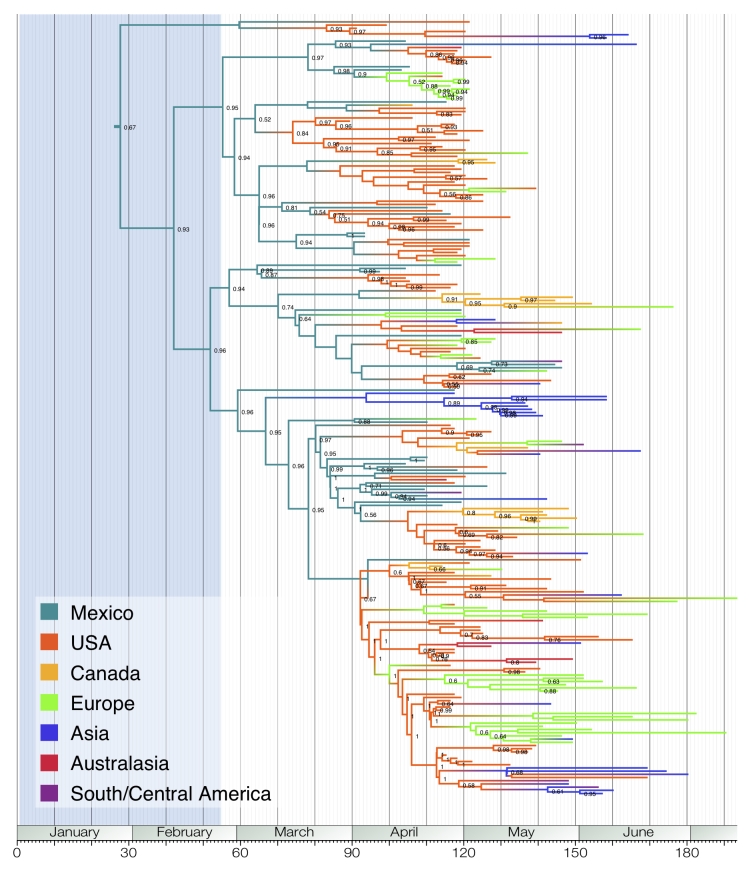
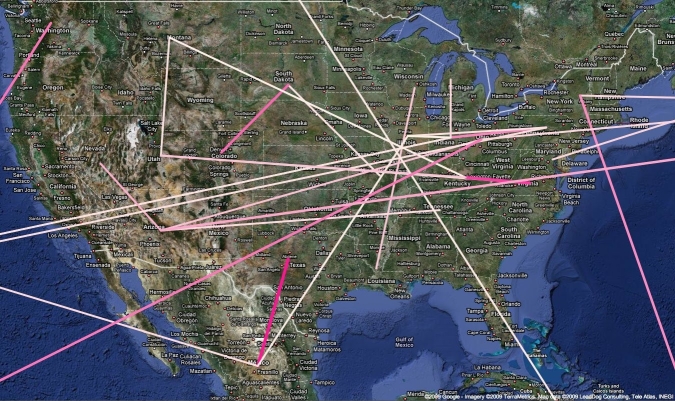
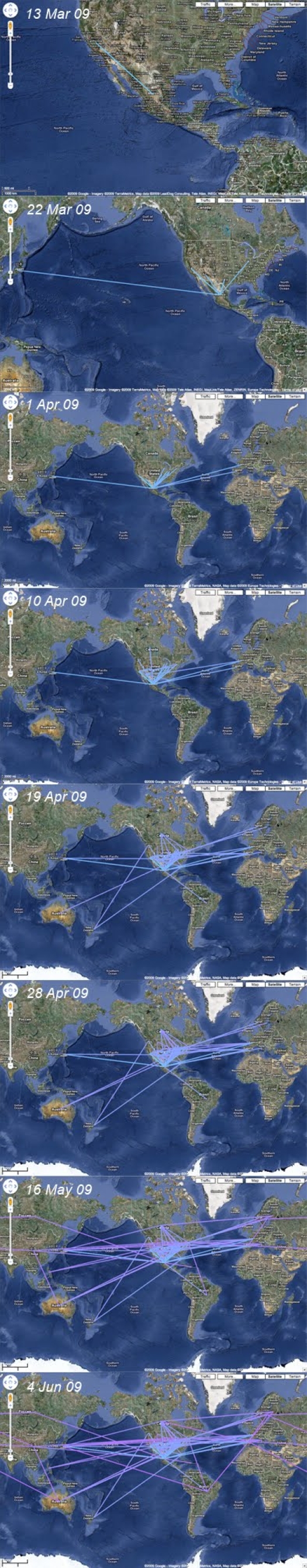
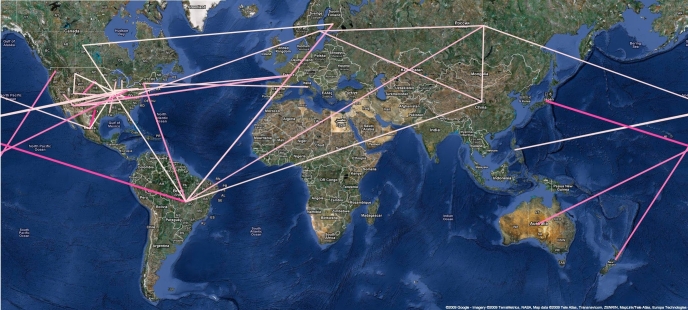
Here, we present an analysis of the H1N1pdm genetic data sampled over the initial stages in the epidemic. To infer phylodynamic spread in time and space we employ a recently developed Bayesian statistical inference framework (Lemey et al., in press). We model spatial diffusion as a continuous-time Markov chain process along time-measured genealogies. In this analysis, we consider 40 locations for which sequence data were available on 06-Aug-2009. The sampling time interval of the 242 sequences spans from 30-Mar-2009 to 12-Jul-2009. The Bayesian inference typically results in a posterior distribution of phylogenetic trees, each having an estimate of the epidemic locations at the ancestral nodes in the tree. We summarize these trees using the most representative clustering pattern and annotate these clusters with the most probable location states. We can visualize this information as tree that grows over time, seeding locations each time an ancestral node is inferred to exist at a different location. A Bayes factor test provides statistical support for epidemiological linkage throughout the evolutionary history. We demonstrate how our full probabilistic approach efficiently tracks an epidemic based on viral genetic data as it unfolds across the globe.
在此,我们展示了对甲型H1N1流感大流行初期所采集的基因数据的分析。为了推断疫情在时间和空间上的系统动力学传播情况,我们采用了一种最近开发的贝叶斯统计推断框架(Lemey等人,即将发表)。我们将空间扩散建模为沿着时间测量的系统发育树的连续时间马尔可夫链过程。在本分析中,我们考虑了40个地点,在2009年8月6日有这些地点的序列数据。242个序列的采样时间间隔从2009年3月30日至2009年7月12日。贝叶斯推断通常会产生系统发育树的后验分布,每棵树在树的祖先节点处都有疫情地点的估计值。我们使用最具代表性的聚类模式来总结这些树,并用最可能的地点状态标注这些聚类。我们可以将此信息可视化为一棵随时间生长的树,每当推断出一个祖先节点存在于不同地点时,就标记出种子地点。贝叶斯因子检验为整个进化历史中的流行病学联系提供了统计支持。我们展示了我们的全概率方法如何基于病毒基因数据在全球范围内展开时有效地追踪疫情。