Laboratório de Endocrinologia Celular e Molecular LIM-25, Faculdade de Medicina da Universidade de São Paulo, Dr Arnaldo Street, 455, room 4305, São Paulo, Brasil.
BMC Med Genet. 2010 Jan 5;11:3. doi: 10.1186/1471-2350-11-3.
Glycogen storage disease type 0 is an autosomal recessive disease presenting in infancy or early childhood and characterized by ketotic hypoglycemia after prolonged fasting and postprandial hyperglycemia and hyperlactatemia. Sixteen different mutations have been identified to date in the gene which encodes hepatic glycogen synthase, resulting in reduction of glycogen storage in the liver.
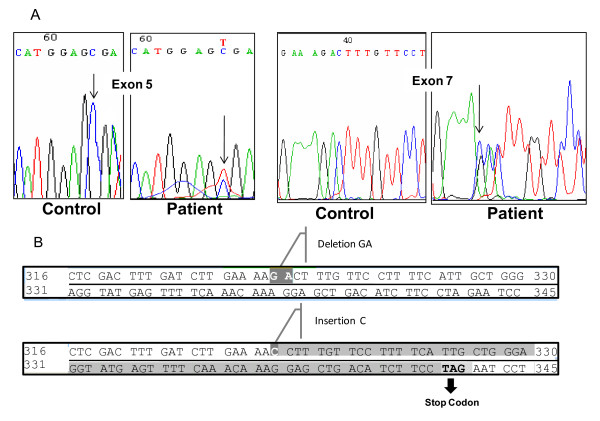
Biochemical evaluation as well as direct sequencing of exons and exon-intron boundary regions of the GYS2 gene were performed in a patient presenting fasting hypoglycemia and postprandial hyperglycemia and her parents. The patient was found to be compound heterozygous for one previously reported nonsense mutation (c.736 C>T; R243X) and a novel frameshift mutation (966_967delGA/insC) which introduces a stop codon 21 aminoacids downstream from the site of the mutation that presumably leads to loss of 51% of the COOH-terminal part of the protein. The glycemia and lactatemia of the parents after an oral glucose tolerance test were evaluated to investigate a possible impact of the carrier status on the metabolic profile. The mother, who presented a positive family history of type 2 diabetes, was classified as glucose intolerant and the father, who did not exhibit metabolic changes after the glucose overload, had an antecedent history of hypoglycemia after moderate alcohol ingestion.
The current results expand the spectrum of known mutations in GYS2 and suggest that haploinsufficiency could explain metabolic abnormalities in heterozygous carriers in presence of predisposing conditions.
糖原贮积病 0 型是一种常染色体隐性疾病,在婴儿期或幼儿期发病,其特征是长时间禁食后出现酮症性低血糖、餐后高血糖和高乳酸血症。迄今为止,已在编码肝糖原合酶的基因中发现了 16 种不同的突变,导致肝脏糖原储存减少。
对表现为空腹低血糖和餐后高血糖的患者及其父母进行了生化评估以及 GYS2 基因外显子和外显子-内含子边界区域的直接测序。患者被发现为复合杂合子,携带一种先前报道的无义突变(c.736C>T;R243X)和一种新的移码突变(966_967delGA/insC),该突变导致在突变位点下游 21 个氨基酸处引入一个终止密码子,推测导致蛋白羧基末端部分缺失 51%。对父母口服葡萄糖耐量试验后的血糖和血乳酸进行评估,以调查携带者状态对代谢谱的可能影响。母亲有 2 型糖尿病阳性家族史,被归类为葡萄糖不耐受;父亲在葡萄糖负荷后没有代谢变化,但有中度饮酒后低血糖的既往史。
目前的结果扩展了 GYS2 中已知突变的范围,并表明单倍不足可能解释在易感条件下杂合子携带者的代谢异常。