David R, Cheriton School of Computer Science, University of Waterloo, Waterloo, ON N2L 3G1, Canada.
BMC Bioinformatics. 2010 Jan 13;11:25. doi: 10.1186/1471-2105-11-25.
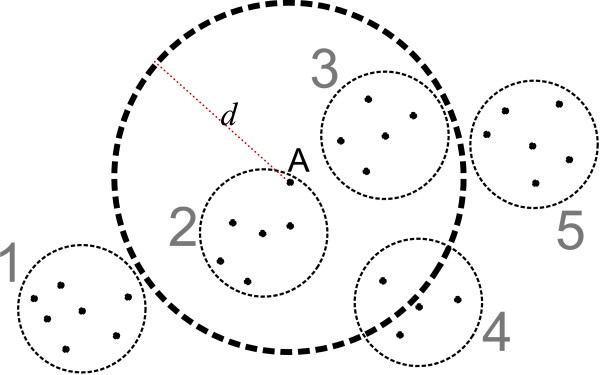
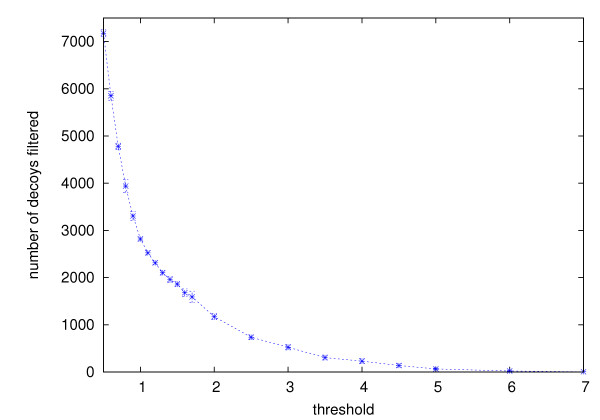
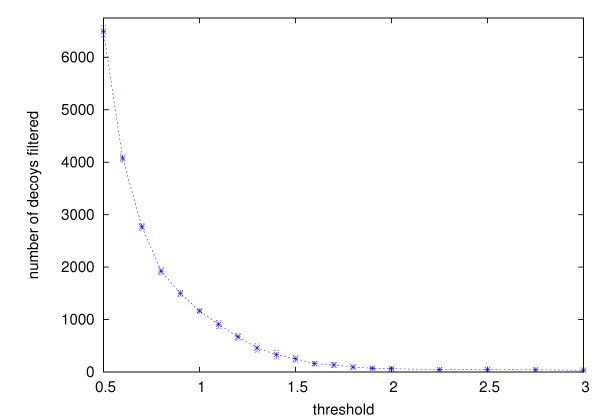
Ab initio protein structure prediction methods generate numerous structural candidates, which are referred to as decoys. The decoy with the most number of neighbors of up to a threshold distance is typically identified as the most representative decoy. However, the clustering of decoys needed for this criterion involves computations with runtimes that are at best quadratic in the number of decoys. As a result currently there is no tool that is designed to exactly cluster very large numbers of decoys, thus creating a bottleneck in the analysis.
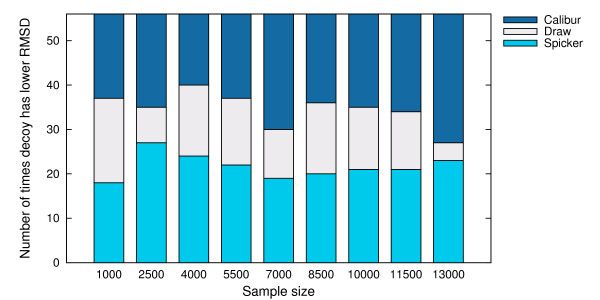
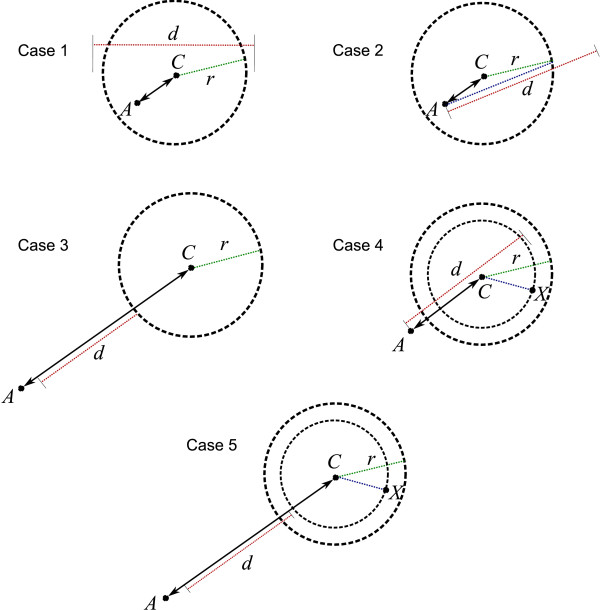
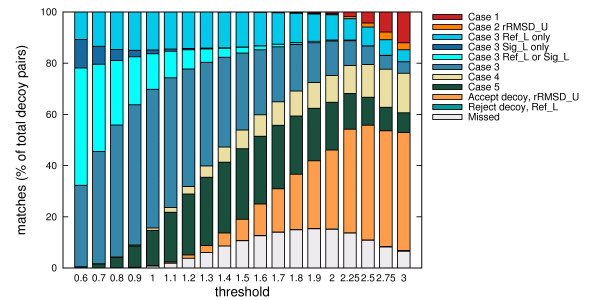
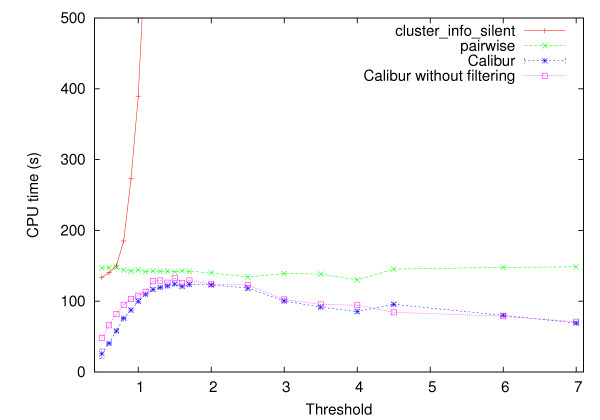
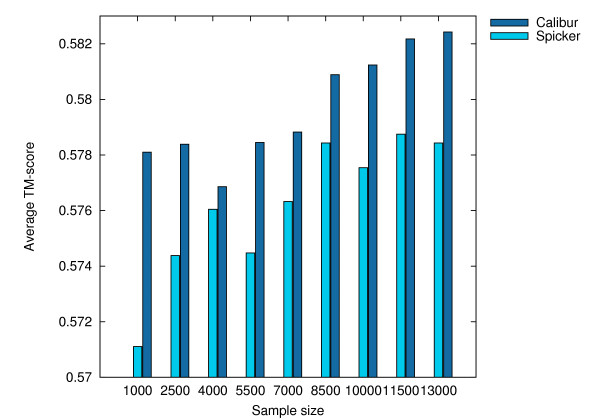
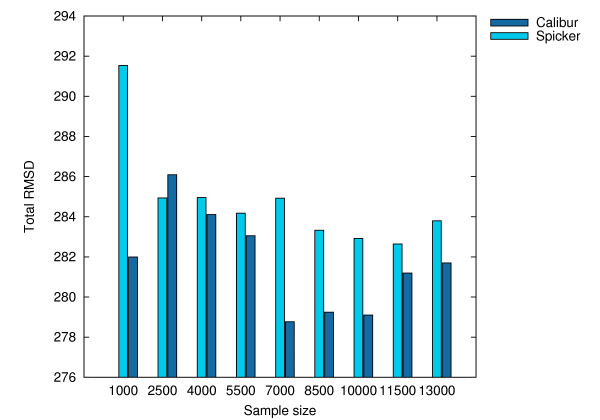
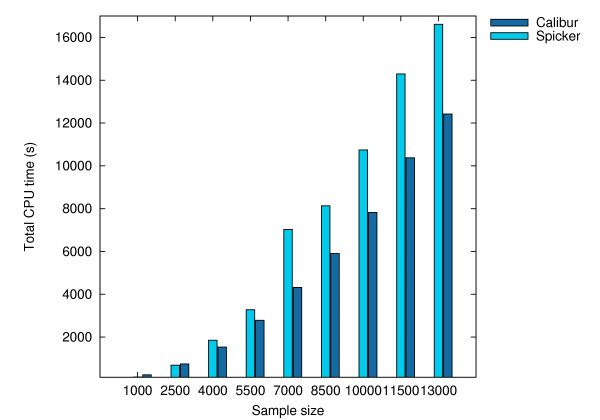
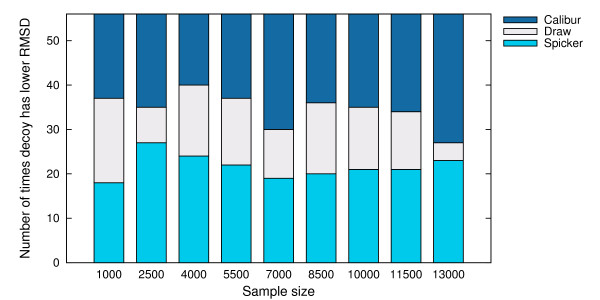
Using three strategies aimed at enhancing performance (proximate decoys organization, preliminary screening via lower and upper bounds, outliers filtering) we designed and implemented a software tool for clustering decoys called Calibur. We show empirical results indicating the effectiveness of each of the strategies employed. The strategies are further fine-tuned according to their effectiveness.Calibur demonstrated the ability to scale well with respect to increases in the number of decoys. For a sample size of approximately 30 thousand decoys, Calibur completed the analysis in one third of the time required when the strategies are not used.For practical use Calibur is able to automatically discover from the input decoys a suitable threshold distance for clustering. Several methods for this discovery are implemented in Calibur, where by default a very fast one is used. Using the default method Calibur reported relatively good decoys in our tests.
Calibur's ability to handle very large protein decoy sets makes it a useful tool for clustering decoys in ab initio protein structure prediction. As the number of decoys generated in these methods increases, we believe Calibur will come in important for progress in the field.
从头蛋白质结构预测方法生成大量的结构候选者,这些候选者被称为诱饵。具有最多邻居数量的诱饵通常被识别为最具代表性的诱饵,这些邻居的数量最多可达一个阈值距离。然而,用于该标准的诱饵聚类涉及到的计算时间在最好的情况下是诱饵数量的二次方。因此,目前没有设计用于精确聚类大量诱饵的工具,从而在分析中形成了瓶颈。
我们使用了三种旨在提高性能的策略(接近诱饵的组织、通过上下界进行初步筛选、异常值过滤),设计并实现了一种称为 Calibur 的诱饵聚类软件工具。我们展示了表明所采用的每种策略的有效性的经验结果。根据其有效性进一步对策略进行微调。Calibur 证明了能够很好地扩展到诱饵数量增加的能力。对于大约 30000 个诱饵的样本量,Calibur 在不使用策略的情况下完成分析所需时间的三分之一。对于实际使用,Calibur 能够自动从输入诱饵中发现适合聚类的合适阈值距离。Calibur 中实现了几种用于此发现的方法,默认使用非常快速的方法。使用默认方法,Calibur 在我们的测试中报告了相对较好的诱饵。
Calibur 能够处理非常大的蛋白质诱饵集,使其成为从头蛋白质结构预测中聚类诱饵的有用工具。随着这些方法生成的诱饵数量的增加,我们相信 Calibur 将对该领域的进展变得非常重要。