Hospital Universitario Central de Asturias, Oviedo, Asturias, Spain.
Orphanet J Rare Dis. 2010 Jan 14;5:1. doi: 10.1186/1750-1172-5-1.
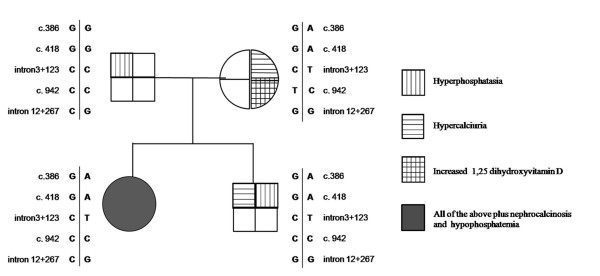
Hereditary hypophosphatemic rickets with hypercalciuria is a rare autosomal recessive disorder (OMIM #241530), characterized by decreased renal phosphate reabsorption that leads to hypophosphatemia, rickets, and bone pain; hypophosphatemia is believed to stimulate 1,25 dihydroxyvitamin D synthesis which, in turn, results in hypercalciuria. Hereditary hypophosphatemic rickets with hypercalciuria is caused by loss-of-function in the type 2c sodium phosphate cotransporter encoded by the SLC34A3 gene. This report shows a family with a non-previously identified mutation in the SLC34A3 gene and exhibiting mild and different manifestations of HHRH. The probandus had hypophosphatemia, elevated serum 1,25 dihydroxyvitamin D concentrations, high serum alkaline phosphatase levels, hypercalciuria and nephrocalcinosis. The other members of the family presented some of these alterations: the mother, hypercalciuria and high 1,25 dihydroxyvitamin D concentrations; the son, hypercalciuria, high 1,25 dihydroxyvitamin D values and elevated alkaline phosphatases; the father, high alkaline phosphatases. The genetic analysis revealed the existence of a single mutation (G78R) in heterozygosis in the SLC34A3 gene in the probandus, her mother and her brother, but not in the father. These findings suggest that he mutation in heterozygosis likely gave rise to a mild phenotype with different penetrance in the three relatives and also indicates that the elevation of 1,25 dihydroxyvitamin D does not result from hypophosphatemia. Thus, this family raises some issues on the transmission and pathophysiology of hereditary hypophosphatemic rickets with hypercalciuria.
遗传性低血磷性抗维生素 D 佝偻病伴高钙尿症是一种罕见的常染色体隐性遗传疾病(OMIM#241530),其特征为肾脏磷酸盐重吸收减少导致低血磷症、佝偻病和骨痛;低血磷症被认为会刺激 1,25 二羟维生素 D 的合成,进而导致高钙尿症。遗传性低血磷性抗维生素 D 佝偻病伴高钙尿症是由 SLC34A3 基因编码的 2c 型钠磷酸盐协同转运蛋白功能丧失引起的。本报告展示了一个家族,该家族中 SLC34A3 基因存在一个先前未识别的突变,表现出轻度和不同表现的 HHRH。先证者存在低血磷症、血清 1,25 二羟维生素 D 浓度升高、血清碱性磷酸酶水平升高、高钙尿症和肾钙质沉着症。家族中的其他成员表现出其中一些改变:母亲表现为高钙尿症和高 1,25 二羟维生素 D 浓度;儿子表现为高钙尿症、高 1,25 二羟维生素 D 值和升高的碱性磷酸酶;父亲表现为高碱性磷酸酶。基因分析显示,先证者、其母亲和其兄弟均存在 SLC34A3 基因杂合单个突变(G78R),但父亲没有。这些发现表明,杂合突变可能导致了这三个亲属中具有不同外显率的轻度表型,也表明 1,25 二羟维生素 D 的升高并非由低血磷症引起。因此,该家族提出了遗传性低血磷性抗维生素 D 佝偻病伴高钙尿症在遗传和病理生理学方面的一些问题。