Ross 858, Division of Cardiology, Department of Medicine, Johns Hopkins University Medical Institutions, Baltimore, MD, 21205, USA.
Basic Res Cardiol. 2010 May;105(3):337-47. doi: 10.1007/s00395-010-0084-5. Epub 2010 Jan 27.
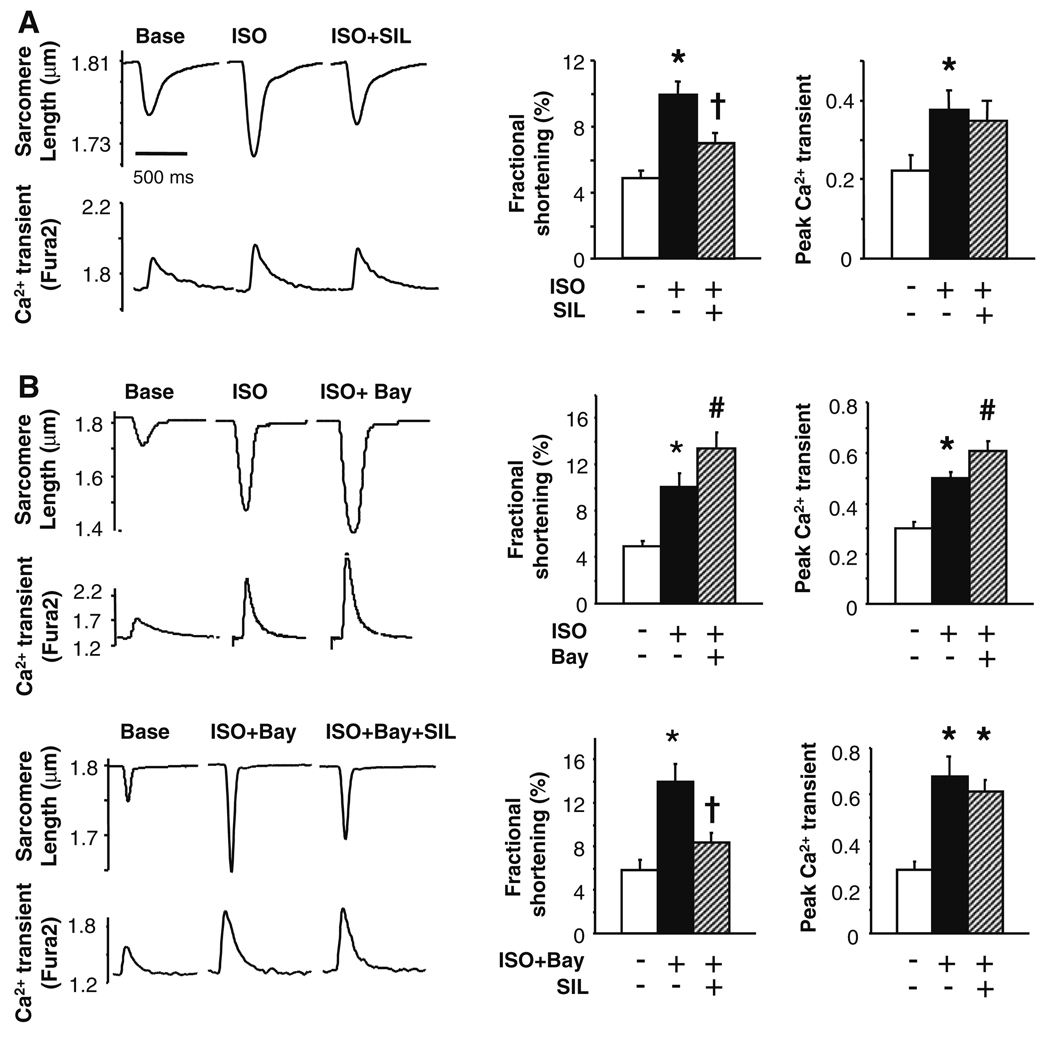
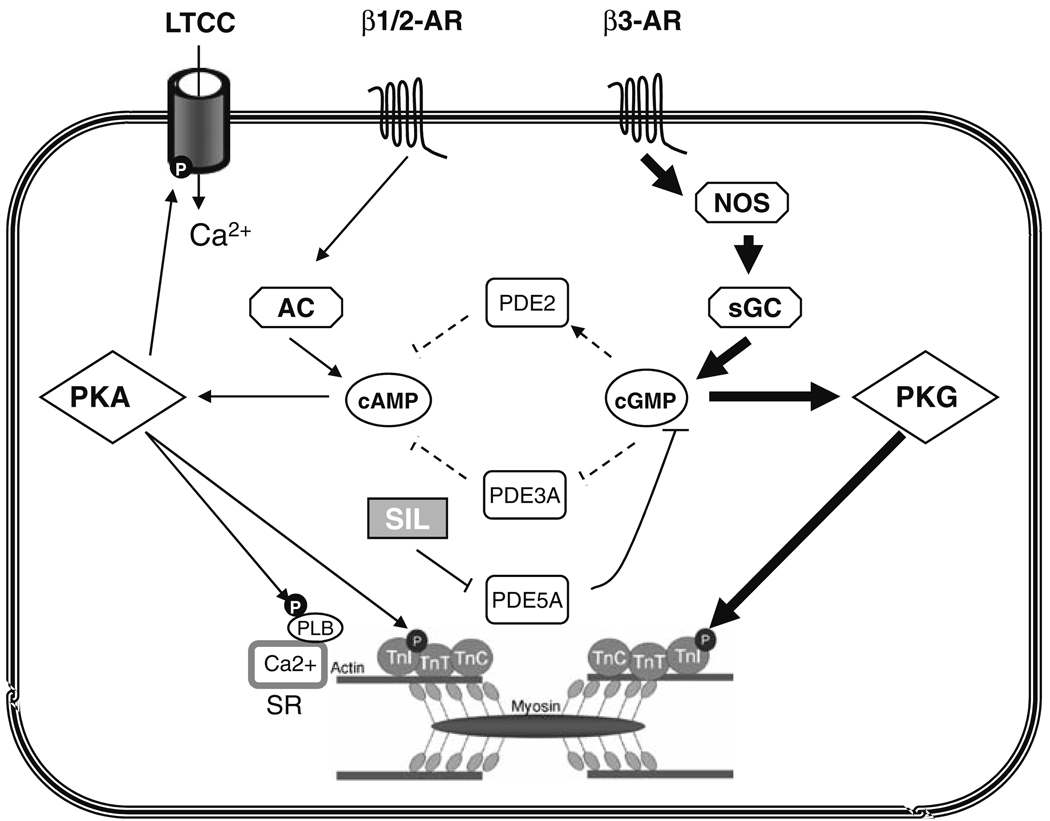
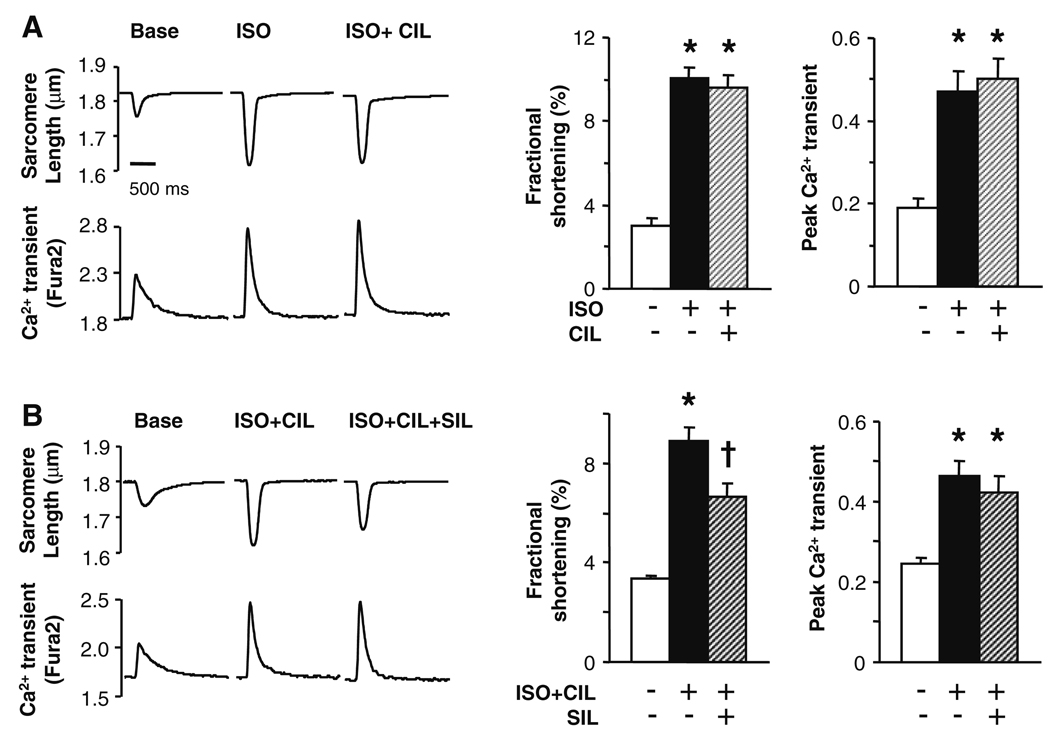
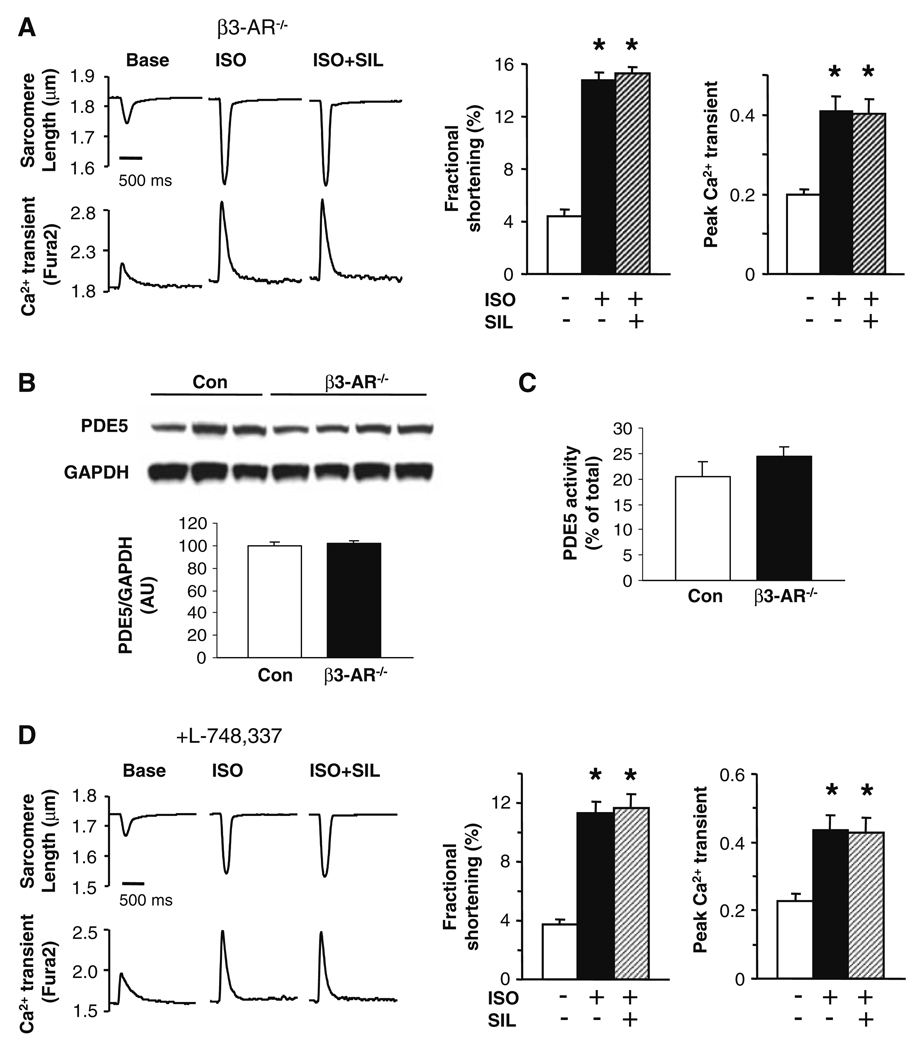
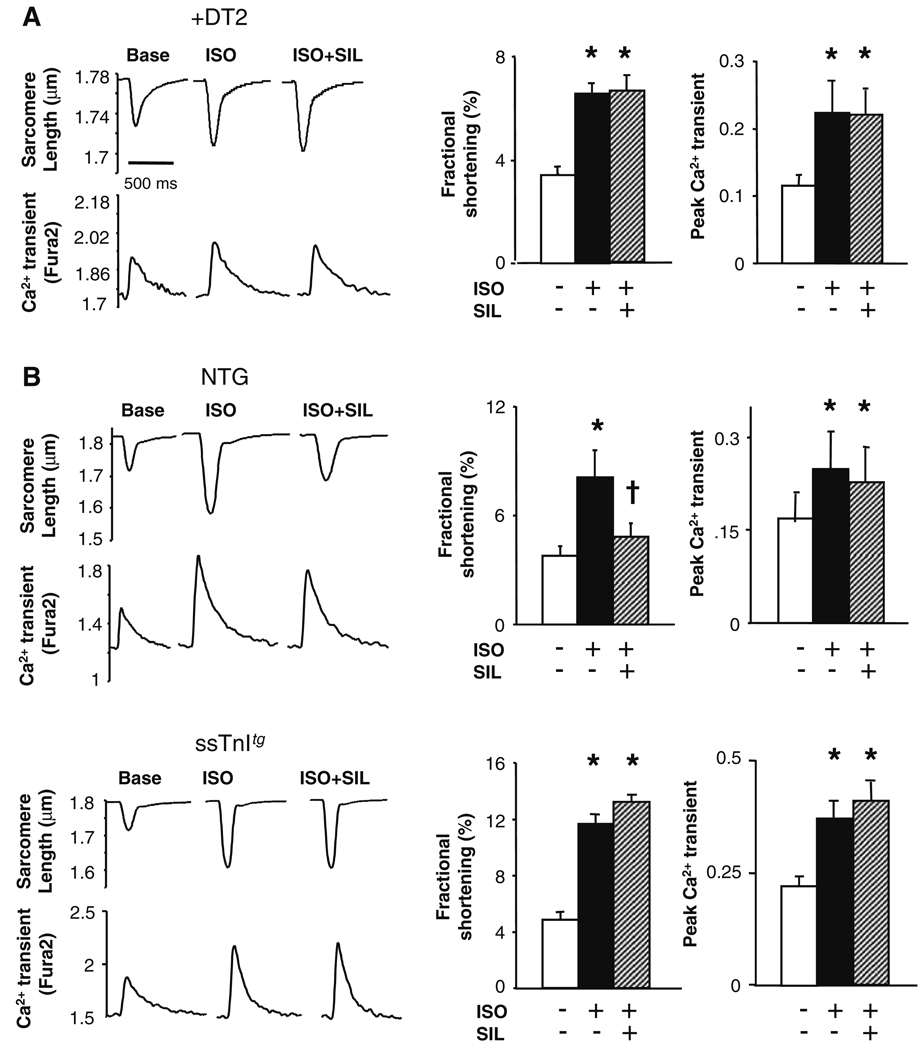
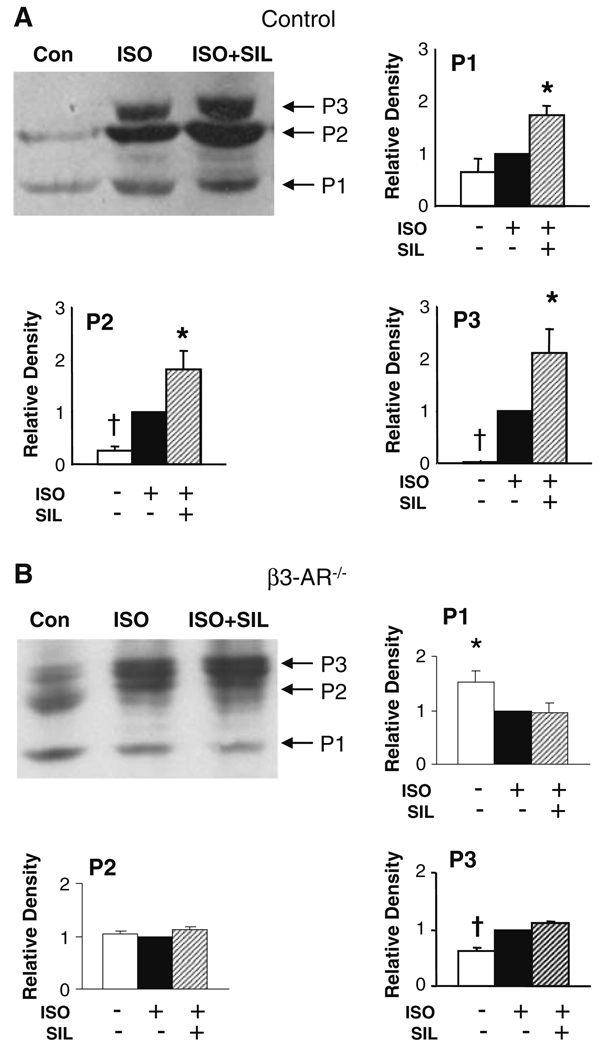
Phosphodiesterase type 5A (PDE5A) inhibitors acutely suppress beta-adrenergic receptor (beta-AR) stimulation in left ventricular myocytes and hearts. This modulation requires cyclic GMP synthesis via nitric oxide synthase (NOS)-NO stimulation, but upstream and downstream mechanisms remain un-defined. To determine this, adult cardiac myocytes from genetically engineered mice and controls were studied by video microscopy to assess sarcomere shortening (SS) and fura2-AM fluorescence to measure calcium transients (CaT). Enhanced SS from isoproterenol (ISO, 10 nM) was suppressed >or=50% by the PDE5A inhibitor sildenafil (SIL, 1 microM), without altering CaT. This regulation was unaltered despite co-inhibition of either the cGMP-stimulated cAMP-esterase PDE2 (Bay 60-7550), or cGMP-inhibited cAMP-esterase PDE3 (cilostamide). Thus, the SIL response could not be ascribed to cGMP interaction with alternative PDEs. However, genetic deletion (or pharmacologic blockade) of beta3-ARs, which couple to NOS signaling, fully prevented SIL modulation of ISO-stimulated SS. Importantly, both PDE5A protein expression and activity were similar in beta3-AR knockout (beta3-AR(-/-)) myocytes as in controls. Downstream, cGMP stimulates protein kinase G (PKG), and we found contractile modulation by SIL required PKG activation and enhanced TnI phosphorylation at S23, S24. Myocytes expressing the slow skeletal TnI isoform which lacks these sites displayed no modulation of ISO responses by SIL. Non-equilibrium isoelectric focusing gel electrophoresis showed SIL increased TnI phosphorylation above that from concomitant ISO in control but not beta3-AR(-/-) myocytes. These data support a cascade involving beta3-AR stimulation, and subsequent PKG-dependent TnI S23, S24 phosphorylation as primary factors underlying the capacity of acute PDE5A inhibition to blunt myocardial beta-adrenergic stimulation.
磷酸二酯酶 5A(PDE5A)抑制剂可急性抑制左心室心肌细胞和心脏中的β肾上腺素能受体(β-AR)刺激。这种调节需要通过一氧化氮合酶(NOS)-NO 刺激合成环鸟苷酸(cGMP),但上游和下游机制仍未确定。为了确定这一点,通过视频显微镜研究了来自基因工程小鼠和对照的成年心肌细胞,以评估肌节缩短(SS)和 fura2-AM 荧光测量钙瞬变(CaT)。PDE5A 抑制剂西地那非(SIL,1 μM)抑制异丙肾上腺素(ISO,10 nM)引起的 SS 增强> = 50%,而不改变 CaT。尽管同时抑制 cGMP 刺激的 cAMP-酯酶 PDE2(Bay 60-7550)或 cGMP 抑制的 cAMP-酯酶 PDE3(西洛他唑),这种调节也没有改变。因此,SIL 的反应不能归因于 cGMP 与替代 PDE 的相互作用。然而,β3-AR 的基因缺失(或药理学阻断),其与 NOS 信号传导偶联,完全阻止了 SIL 对 ISO 刺激的 SS 的调节。重要的是,β3-AR 敲除(β3-AR(-/-))心肌细胞中的 PDE5A 蛋白表达和活性与对照相似。下游,cGMP 刺激蛋白激酶 G(PKG),我们发现 SIL 的收缩调节需要 PKG 激活和增强 TnI 在 S23、S24 的磷酸化。表达缺乏这些位点的慢骨骼肌 TnI 同工型的心肌细胞对 SIL 没有调节 ISO 反应。非平衡等电聚焦凝胶电泳显示,SIL 增加了 TnI 的磷酸化,超过了对照心肌细胞中同时存在的 ISO 的磷酸化,但在β3-AR(-/-)心肌细胞中则没有。这些数据支持了一个级联反应,涉及β3-AR 刺激,以及随后 PKG 依赖性 TnI S23、S24 磷酸化,作为急性 PDE5A 抑制抑制心肌β肾上腺素刺激能力的主要因素。