Department of Cardiology and Angiology, University Hospital Münster, Albert-Schweitzer-Street 33, D-48129 Münster, Germany.
Cardiovasc Res. 2010 Jul 1;87(1):60-72. doi: 10.1093/cvr/cvq029. Epub 2010 Jan 28.
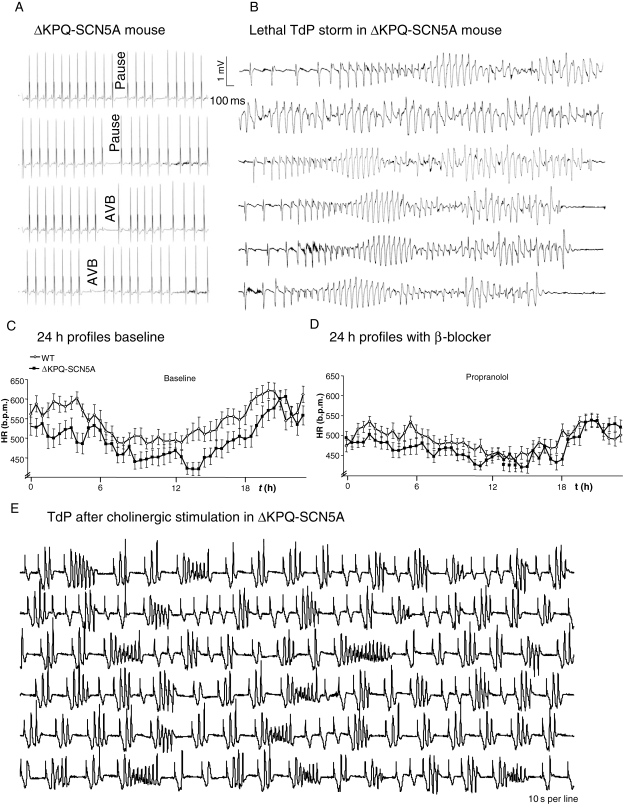
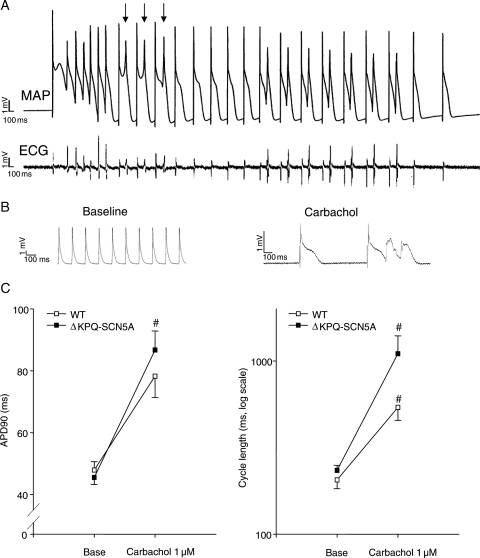
Clinical observations in patients with long QT syndrome carrying sodium channel mutations (LQT3) suggest that bradycardia caused by parasympathetic stimulation may provoke torsades de pointes (TdP). Beta-adrenoceptor blockers appear less effective in LQT3 than in other forms of the disease.
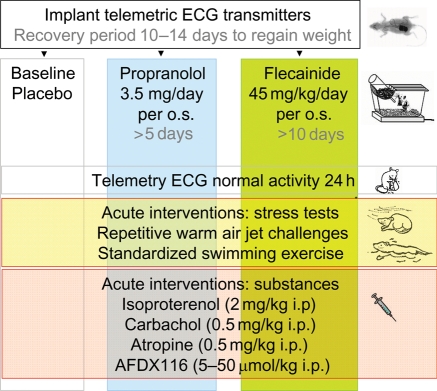
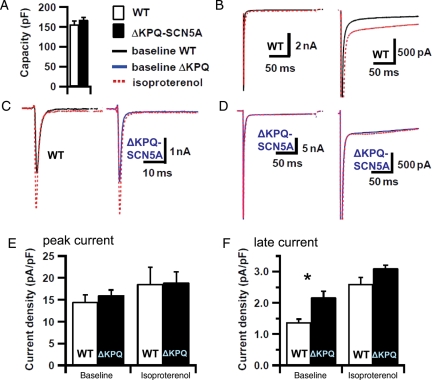
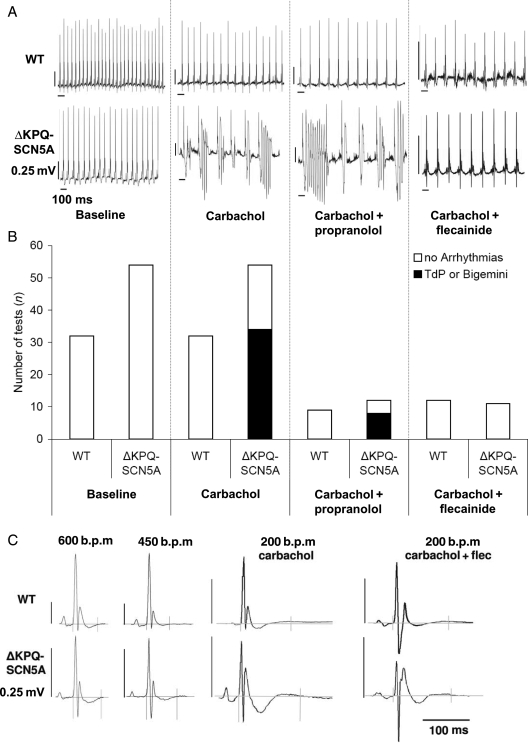
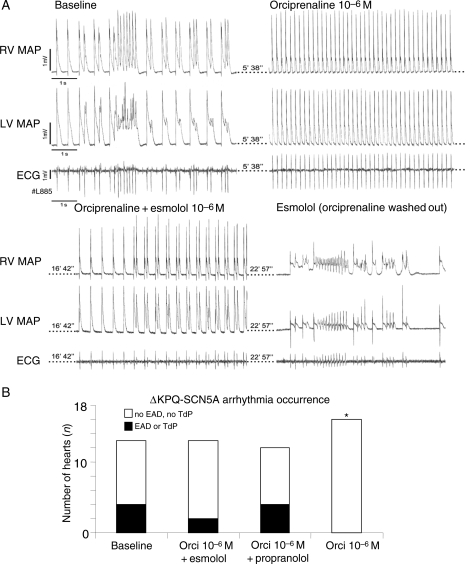
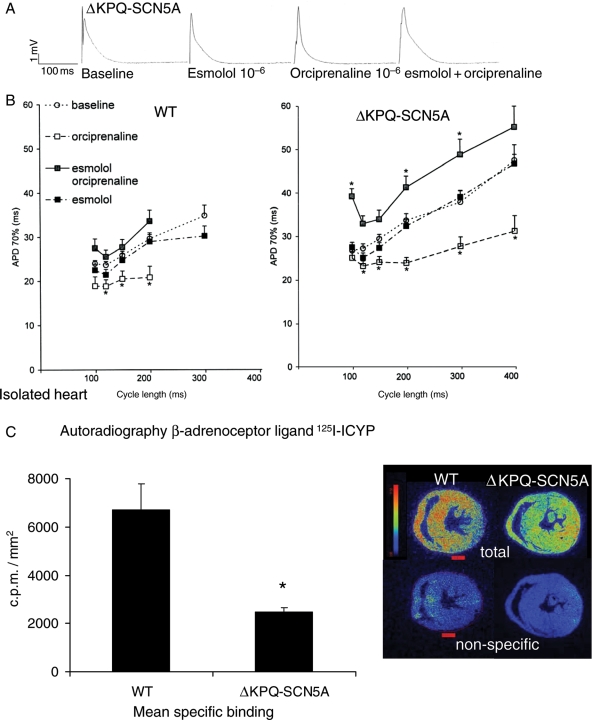
We studied effects of autonomic modulation on arrhythmias in vivo and in vitro and quantified sympathetic innervation by autoradiography in heterozygous mice with a knock-in deletion (DeltaKPQ) in the Scn5a gene coding for the cardiac sodium channel and increased late sodium current (LQT3 mice). Cholinergic stimulation by carbachol provoked bigemini and TdP in freely roaming LQT3 mice. No arrhythmias were provoked by physical stress, mental stress, isoproterenol, or atropine. In isolated, beating hearts, carbachol did not prolong action potentials per se, but caused bradycardia and rate-dependent action potential prolongation. The muscarinic inhibitor AFDX116 prevented effects of carbachol on heart rate and arrhythmias. beta-Adrenoceptor stimulation suppressed arrhythmias, shortened rate-corrected action potential duration, increased rate, and minimized difference in late sodium current between genotypes. Beta-adrenoceptor density was reduced in LQT3 hearts. Acute beta-adrenoceptor blockade by esmolol, propranolol or chronic propranolol in vivo did not suppress arrhythmias. Chronic flecainide pre-treatment prevented arrhythmias (all P < 0.05).
Cholinergic stimulation provokes arrhythmias in this model of LQT3 by triggering bradycardia. beta-Adrenoceptor density is reduced, and beta-adrenoceptor blockade does not prevent arrhythmias. Sodium channel blockade and beta-adrenoceptor stimulation suppress arrhythmias by shortening repolarization and minimizing difference in late sodium current.
长 QT 综合征伴有钠离子通道突变(LQT3)患者的临床观察表明,迷走神经刺激引起的心动过缓可能引发尖端扭转型室性心动过速(TdP)。β-肾上腺素受体阻滞剂在 LQT3 中的疗效似乎不如其他形式的疾病。
我们研究了自主神经调节对体内和体外心律失常的影响,并通过放射性自显影技术对编码心脏钠离子通道的 Scn5a 基因中的敲入缺失(DeltaKPQ)的杂合子小鼠进行定量分析,该缺失导致晚期钠离子电流增加(LQT3 小鼠)。卡巴胆碱引起的胆碱能刺激在自由漫游的 LQT3 小鼠中引发二联律和 TdP。身体应激、精神应激、异丙肾上腺素或阿托品均未引起心律失常。在分离的跳动心脏中,卡巴胆碱本身不会延长动作电位,但会导致心动过缓和频率依赖性动作电位延长。毒蕈碱抑制剂 AFDX116 可预防卡巴胆碱对心率和心律失常的影响。β-肾上腺素受体刺激可抑制心律失常,缩短心率校正的动作电位持续时间,增加心率,并使基因型之间晚期钠离子电流的差异最小化。LQT3 心脏中的β-肾上腺素受体密度降低。在体急性β-肾上腺素受体阻断剂(如 esmolol、普萘洛尔或慢性普萘洛尔)不能抑制心律失常。慢性氟卡尼预处理可预防心律失常(均 P < 0.05)。
在这种 LQT3 模型中,胆碱能刺激通过引发心动过缓引发心律失常。β-肾上腺素受体密度降低,β-肾上腺素受体阻断不能预防心律失常。钠离子通道阻断和β-肾上腺素受体刺激通过缩短复极和最小化晚期钠离子电流的差异来抑制心律失常。