Department of Information Engineering, University of Padova, Italy.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S16. doi: 10.1186/1471-2105-11-S1-S16.
The classification of protein sequences using string algorithms provides valuable insights for protein function prediction. Several methods, based on a variety of different patterns, have been previously proposed. Almost all string-based approaches discover patterns that are not "independent, " and therefore the associated scores overcount, a multiple number of times, the contribution of patterns that cover the same region of a sequence.
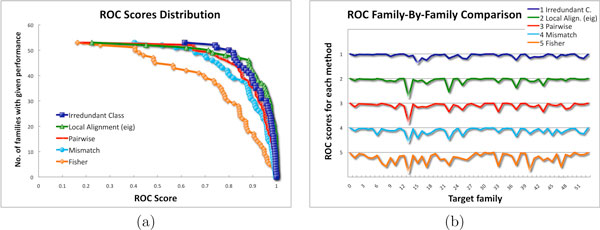
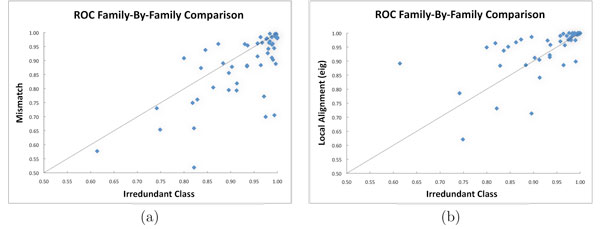
In this paper we use a class of patterns, called irredundant, that is specifically designed to address this issue. Loosely speaking the set of irredundant patterns is the smallest class of "independent" patterns that can describe all common patterns in two sequences, thus they avoid overcounting. We present a novel discriminative method, called Irredundant Class, based on the statistics of irredundant patterns combined with the power of support vector machines.
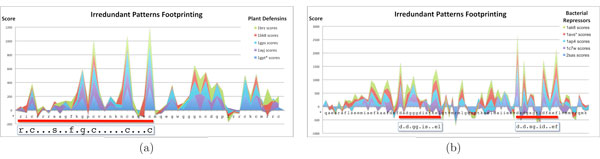
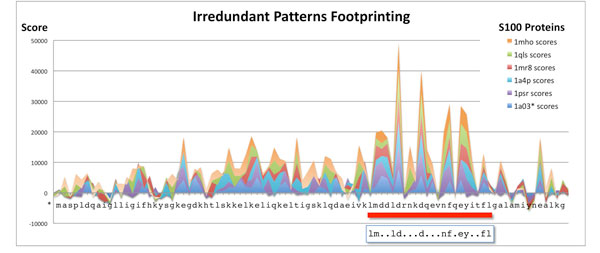
Tests on benchmark data show that Irredundant Class outperforms most of the string algorithms previously proposed, and it achieves results as good as current state-of-the-art methods. Moreover the footprints of the most discriminative irredundant patterns can be used to guide the identification of functional regions in protein sequences.
使用字符串算法对蛋白质序列进行分类为蛋白质功能预测提供了有价值的见解。以前已经提出了几种基于各种不同模式的方法。几乎所有基于字符串的方法都发现了不是“独立”的模式,因此相关的得分多次重复计算了覆盖序列同一区域的模式的贡献。
在本文中,我们使用了一类称为非冗余的模式,专门用于解决这个问题。从广义上讲,非冗余模式集是可以描述两个序列中所有常见模式的“独立”模式的最小集合,因此它们避免了重复计数。我们提出了一种新的有区别的方法,称为非冗余类,它基于非冗余模式的统计数据和支持向量机的威力。
在基准数据上的测试表明,非冗余类优于以前提出的大多数字符串算法,并且它的结果与当前最先进的方法一样好。此外,最具区分性的非冗余模式的足迹可用于指导蛋白质序列中功能区域的识别。