Department of Statistics, Operations, and Management Science, The University of Tennessee, 331 Stokely Management Center, Knoxville, TN 37996, USA.
BMC Bioinformatics. 2010 Feb 3;11:72. doi: 10.1186/1471-2105-11-72.
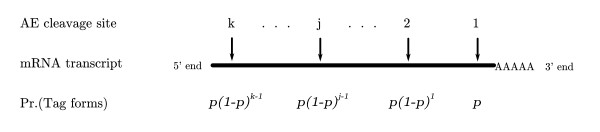
Tag-based techniques, such as SAGE, are commonly used to sample the mRNA pool of an organism's transcriptome. Incomplete digestion during the tag formation process may allow for multiple tags to be generated from a given mRNA transcript. The probability of forming a tag varies with its relative location. As a result, the observed tag counts represent a biased sample of the actual transcript pool. In SAGE this bias can be avoided by ignoring all but the 3' most tag but will discard a large fraction of the observed data. Taking this bias into account should allow more of the available data to be used leading to increased statistical power.
Three new hierarchical models, which directly embed a model for the variation in tag formation probability, are proposed and their associated Bayesian inference algorithms are developed. These models may be applied to libraries at both the tag and aggregate level. Simulation experiments and analysis of real data are used to contrast the accuracy of the various methods. The consequences of tag formation bias are discussed in the context of testing differential expression. A description is given as to how these algorithms can be applied in that context.
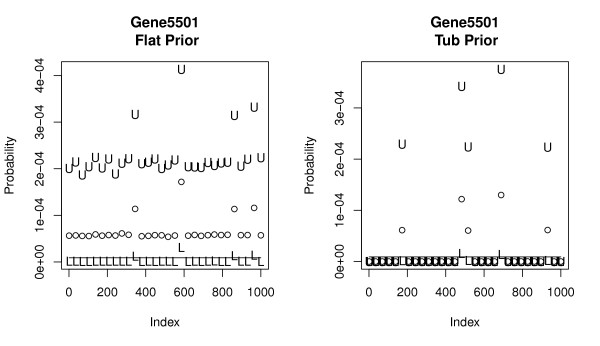
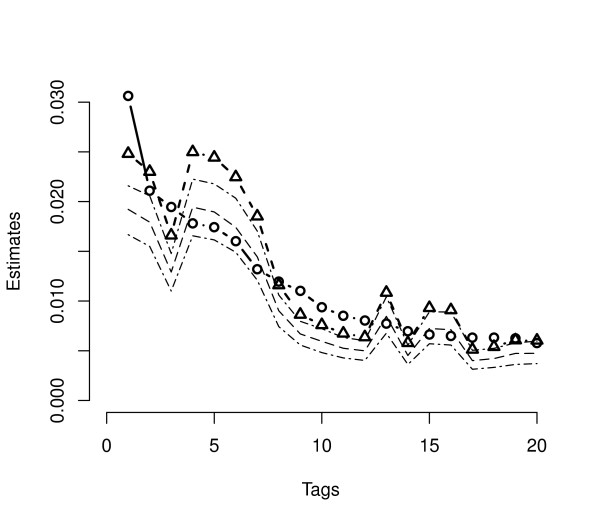
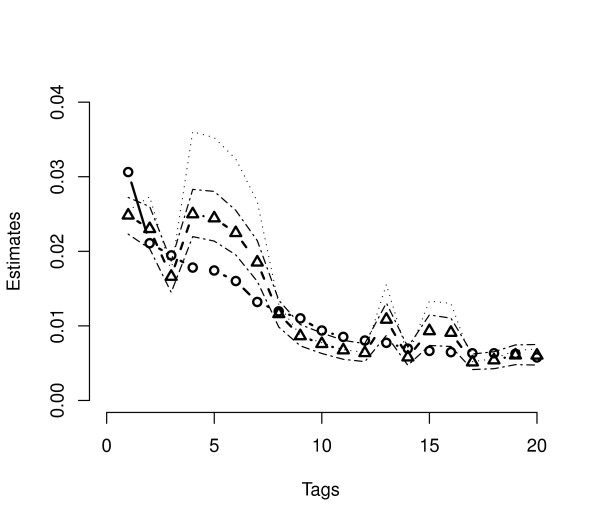
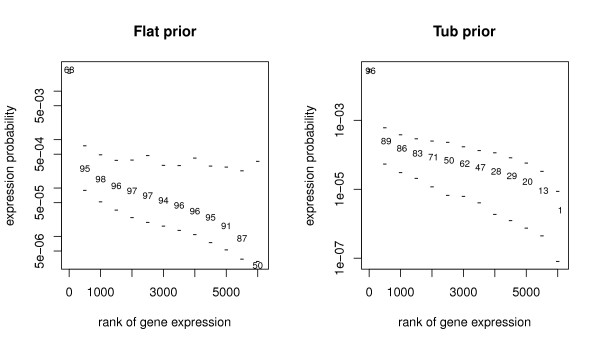
Several Bayesian inference algorithms that account for tag formation effects are compared with the DPB algorithm providing clear evidence of superior performance. The accuracy of inferences when using a particular non-informative prior is found to depend on the expression level of a given gene. The multivariate nature of the approach easily allows both univariate and joint tests of differential expression. Calculations demonstrate the potential for false positive and negative findings due to variation in tag formation probabilities across samples when testing for differential expression.
基于标签的技术,如 SAGE,常用于对生物体转录组的 mRNA 池进行采样。在标签形成过程中,如果不完全消化,可能会从给定的 mRNA 转录本生成多个标签。形成标签的概率随其相对位置而变化。因此,观察到的标签计数代表实际转录本池的有偏差样本。在 SAGE 中,可以通过忽略除 3'端最接近的标签之外的所有标签来避免这种偏差,但会丢弃大量观察到的数据。考虑到这种偏差,可以使用更多可用数据,从而提高统计能力。
提出了三个新的层次模型,它们直接嵌入了标签形成概率变化的模型,并开发了相应的贝叶斯推断算法。这些模型可应用于标签和聚合水平的文库。通过模拟实验和真实数据的分析,比较了各种方法的准确性。在测试差异表达的背景下,讨论了标签形成偏差的后果。给出了如何在这种情况下应用这些算法的说明。
与 DPB 算法相比,比较了几种考虑标签形成效应的贝叶斯推断算法,清楚地证明了它们具有优越的性能。在使用特定非信息先验时进行推断的准确性取决于给定基因的表达水平。该方法的多元性质很容易允许对差异表达进行单变量和联合检验。计算表明,在测试差异表达时,由于标签形成概率在样本之间的变化,可能会导致假阳性和假阴性发现。