Carels Nicolas, Frías Diego
Fundação Oswaldo Cruz (FIOCRUZ), Instituto Oswaldo Cruz (IOC), Laboratório de Genômica Funcional e Bioinformática, Rio de Janeiro, RJ, Brazil.
Bioinform Biol Insights. 2009 Oct 28;3:141-54. doi: 10.4137/bbi.s3030.
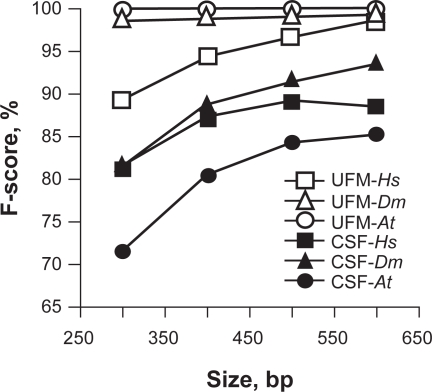
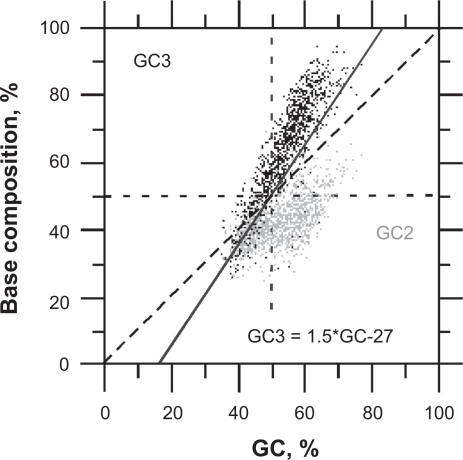
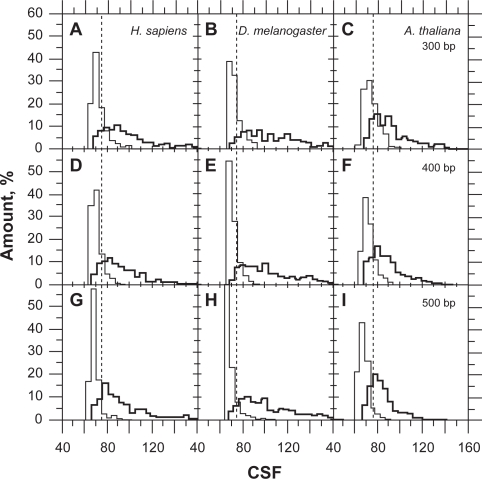
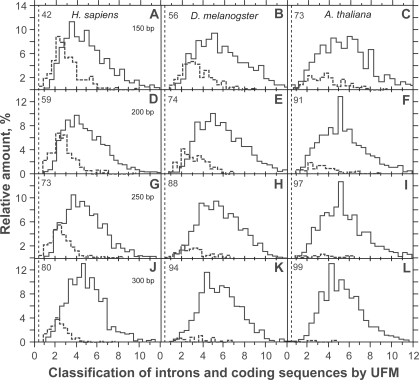
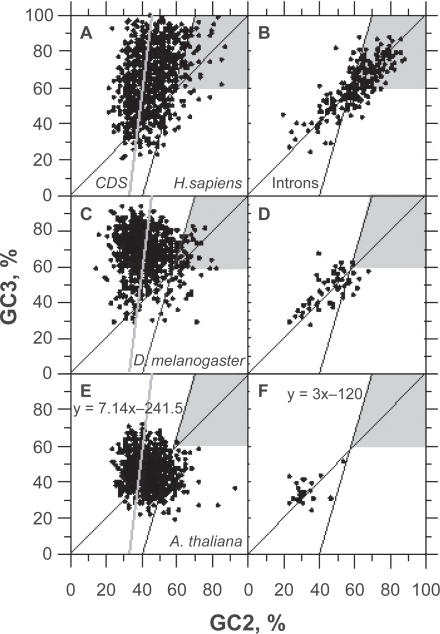
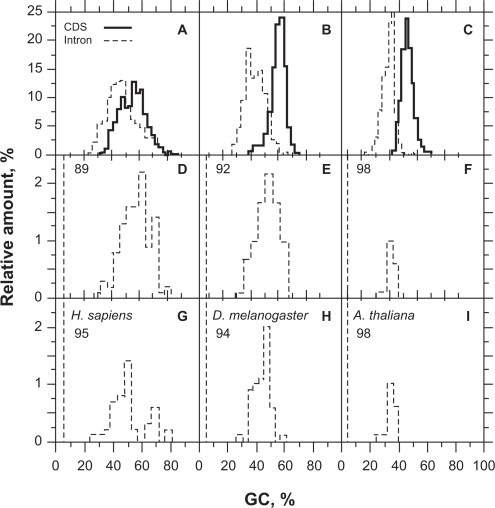
In this report, we compared the success rate of classification of coding sequences (CDS) vs. introns by Codon Structure Factor (CSF) and by a method that we called Universal Feature Method (UFM). UFM is based on the scoring of purine bias (Rrr) and stop codon frequency. We show that the success rate of CDS/intron classification by UFM is higher than by CSF. UFM classifies ORFs as coding or non-coding through a score based on (i) the stop codon distribution, (ii) the product of purine probabilities in the three positions of nucleotide triplets, (iii) the product of Cytosine (C), Guanine (G), and Adenine (A) probabilities in the 1st, 2nd, and 3rd positions of triplets, respectively, (iv) the probabilities of G in 1st and 2nd position of triplets and (v) the distance of their GC3 vs. GC2 levels to the regression line of the universal correlation. More than 80% of CDSs (true positives) of Homo sapiens (>250 bp), Drosophila melanogaster (>250 bp) and Arabidopsis thaliana (>200 bp) are successfully classified with a false positive rate lower or equal to 5%. The method releases coding sequences in their coding strand and coding frame, which allows their automatic translation into protein sequences with 95% confidence. The method is a natural consequence of the compositional bias of nucleotides in coding sequences.
在本报告中,我们比较了密码子结构因子(CSF)和我们称为通用特征法(UFM)的方法对编码序列(CDS)与内含子的分类成功率。UFM基于嘌呤偏好性(Rrr)评分和终止密码子频率。我们表明,UFM对CDS/内含子的分类成功率高于CSF。UFM通过基于以下因素的评分将开放阅读框(ORF)分类为编码或非编码:(i)终止密码子分布,(ii)核苷酸三联体三个位置上嘌呤概率的乘积,(iii)三联体第一、第二和第三位置上胞嘧啶(C)、鸟嘌呤(G)和腺嘌呤(A)概率的乘积,(iv)三联体第一和第二位置上G的概率,以及(v)其GC3与GC2水平到通用相关性回归线的距离。人类(>250 bp)、黑腹果蝇(>250 bp)和拟南芥(>200 bp)中超过80%的CDS(真阳性)被成功分类,假阳性率低于或等于5%。该方法在其编码链和编码框中释放编码序列,这使得它们能够以95%的置信度自动翻译成蛋白质序列。该方法是编码序列中核苷酸组成偏倚的自然结果。