Department of Genome and Gene Expression Data Analysis, Bioinformatics Institute, 30 Biopolis str, Singapore.
BMC Genomics. 2010 Feb 10;11 Suppl 1(Suppl 1):S12. doi: 10.1186/1471-2164-11-S1-S12.
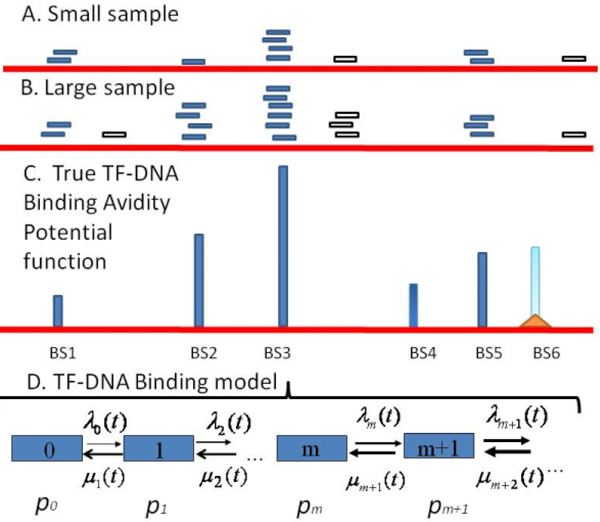
Transcription factor (TF)-DNA binding loci are explored by analyzing massive datasets generated with application of Chromatin Immuno-Precipitation (ChIP)-based high-throughput sequencing technologies. These datasets suffer from a bias in the information about binding loci availability, sample incompleteness and diverse sources of technical and biological noises. Therefore adequate mathematical models of ChIP-based high-throughput assay(s) and statistical tools are required for a robust identification of specific and reliable TF binding sites (TFBS), a precise characterization of TFBS avidity distribution and a plausible estimation the total number of specific TFBS for a given TF in the genome for a given cell type.
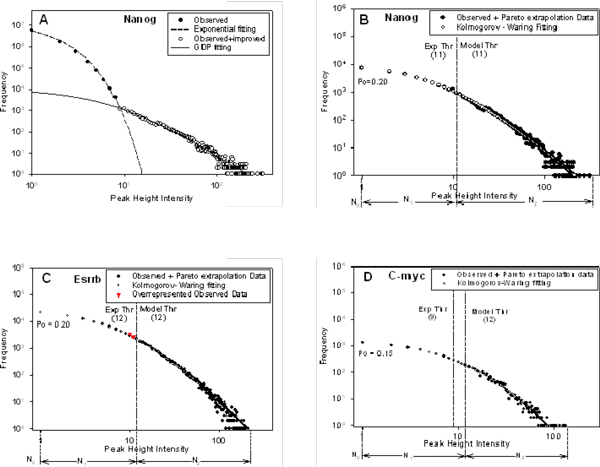
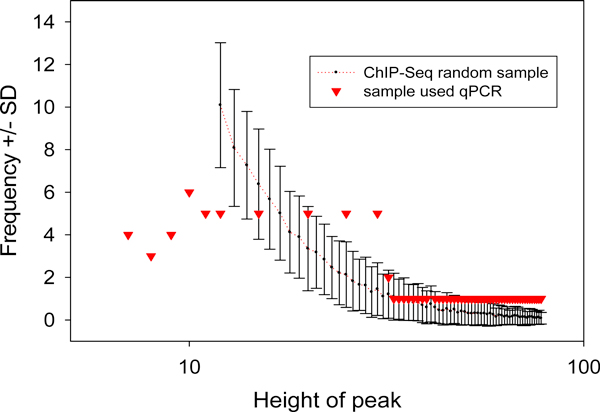
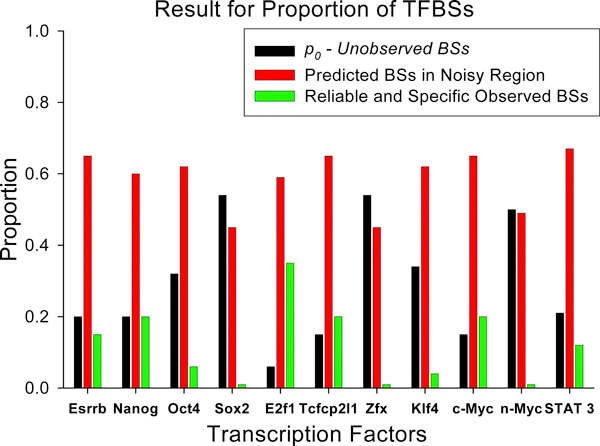
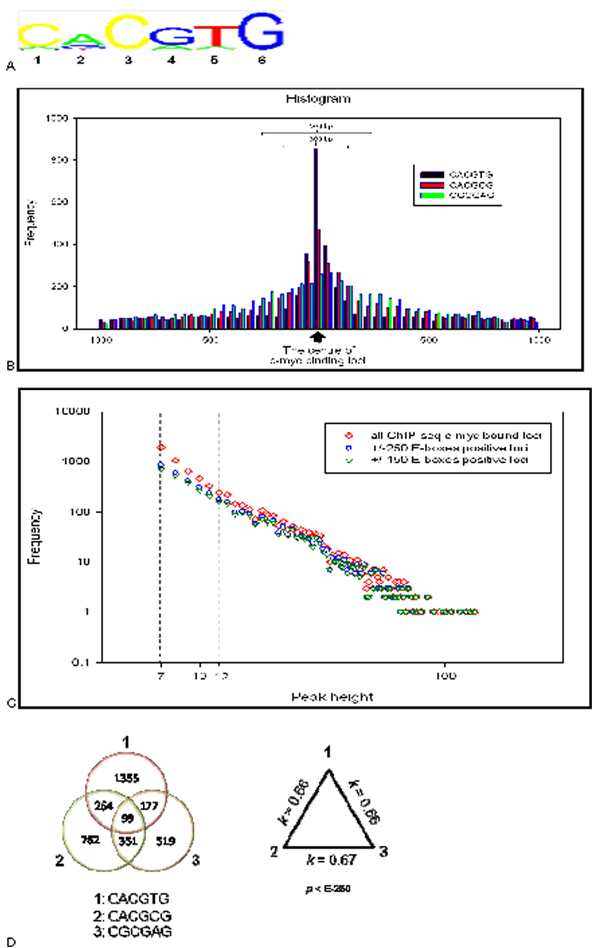
We developed an exploratory mixture probabilistic model for a specific and non-specific transcription factor-DNA (TF-DNA) binding. Within ChiP-seq data sets, the statistics of specific and non-specific DNA-protein binding is defined by a mixture of sample size-dependent skewed functions described by Kolmogorov-Waring (K-W) function (Kuznetsov, 2003) and exponential function, respectively. Using available Chip-seq data for eleven TFs, essential for self-maintenance and differentiation of mouse embryonic stem cells (SC) (Nanog, Oct4, sox2, KLf4, STAT3, E2F1, Tcfcp211, ZFX, n-Myc, c-Myc and Essrb) reported in Chen et al (2008), we estimated (i) the specificity and the sensitivity of the ChiP-seq binding assays and (ii) the number of specific but not identified in the current experiments binding sites (BSs) in the genome of mouse embryonic stem cells. Motif finding analysis applied to the identified c-Myc TFBSs supports our results and allowed us to predict many novel c-Myc target genes.
We provide a novel methodology of estimating the specificity and the sensitivity of TF-DNA binding in massively paralleled ChIP sequencing (ChIP-seq) binding assay. Goodness-of fit analysis of K-W functions suggests that a large fraction of low- and moderate- avidity TFBSs cannot be identified by the ChIP-based methods. Thus the task to identify the binding sensitivity of a TF cannot be technically resolved yet by current ChIP-seq, compared to former experimental techniques. Considering our improvement in measuring the sensitivity and the specificity of the TFs obtained from the ChIP-seq data, the models of transcriptional regulatory networks in embryonic cells and other cell types derived from the given ChIp-seq data should be carefully revised.
转录因子(TF)-DNA 结合位点是通过分析应用染色质免疫沉淀(ChIP)高通量测序技术生成的大量数据集来探索的。这些数据集在绑定位置信息的可用性、样本不完整性以及技术和生物噪声的各种来源方面存在偏差。因此,需要足够的基于 ChIP 的高通量检测的数学模型和统计工具,以稳健地识别特定且可靠的 TF 结合位点(TFBS)、精确表征 TFBS 亲和力分布,并合理估计给定细胞类型中给定 TF 在基因组中的特定 TFBS 的总数。
我们开发了一种用于特定和非特定转录因子-DNA(TF-DNA)结合的探索性混合概率模型。在 ChiP-seq 数据集中,特定和非特定 DNA-蛋白质结合的统计数据由样本大小相关的偏态函数的混合物定义,这些函数由 Kolmogorov-Waring(K-W)函数(Kuznetsov,2003)和指数函数分别描述。使用 Chen 等人(2008 年)报告的用于自我维持和分化的小鼠胚胎干细胞(SC)的 11 种 TF(Nanog、Oct4、sox2、Klf4、STAT3、E2F1、Tcfcp211、ZFX、n-Myc、c-Myc 和 Essrb)的现有 ChiP-seq 数据,我们估计了(i)ChiP-seq 结合测定的特异性和敏感性,以及(ii)在小鼠胚胎干细胞基因组中当前实验无法识别的特定但未识别的结合位点(BS)的数量。应用于鉴定的 c-Myc TFBS 的基序发现分析支持我们的结果,并允许我们预测许多新的 c-Myc 靶基因。
我们提供了一种估计大规模并行 ChIP 测序(ChIP-seq)结合测定中 TF-DNA 结合特异性和敏感性的新方法。K-W 函数的拟合优度分析表明,很大一部分低亲和性和中亲和性 TFBS 无法通过基于 ChIP 的方法识别。因此,与以前的实验技术相比,当前的 ChIP-seq 技术尚无法解决确定 TF 结合敏感性的任务。考虑到我们从 ChiP-seq 数据中提高了测量 TF 的灵敏度和特异性的方法,应该仔细修改从给定 ChIP-seq 数据导出的胚胎细胞和其他细胞类型的转录调控网络模型。