Department of Physics, University of Illinois at Chicago, 60607-7052, USA.
Int J Mol Sci. 2010 Jan 21;11(1):288-303. doi: 10.3390/ijms11010288.
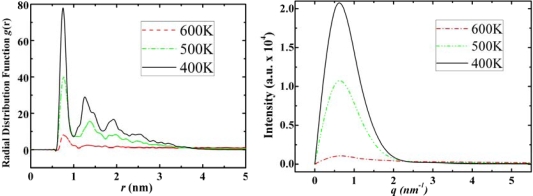
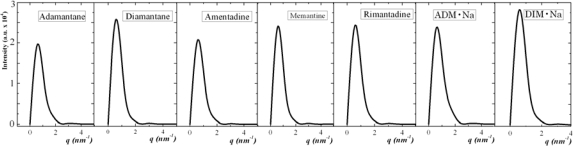
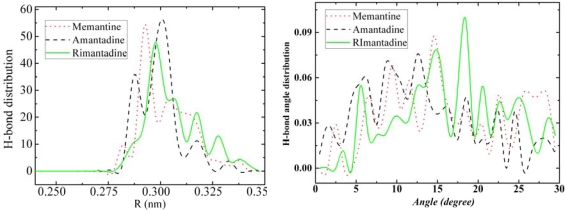
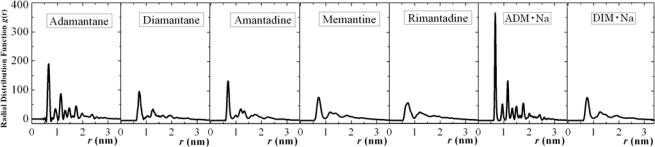
We report self-assembly and phase transition behavior of lower diamondoid molecules and their primary derivatives using molecular dynamics (MD) simulation and density functional theory (DFT) calculations. Two lower diamondoids (adamantane and diamantane), three adamantane derivatives (amantadine, memantine and rimantadine) and two artificial molecules (ADM·Na and DIM·Na) are studied separately in 125-molecule simulation systems. We performed DFT calculations to optimize their molecular geometries and obtained atomic electronic charges for the corresponding MD simulation, by which we predicted self-assembly structures and simulation trajectories for the seven different diamondoids and derivatives. Our radial distribution function and structure factor studies showed clear phase transitions and self-assemblies for the seven diamondoids and derivatives.
我们使用分子动力学(MD)模拟和密度泛函理论(DFT)计算研究了较低的类金刚石分子及其主要衍生物的自组装和相转变行为。我们分别在 125 分子模拟系统中研究了两种较低的类金刚石(金刚烷和二聚金刚烷)、三种金刚烷衍生物(金刚烷胺、美金刚和金刚乙胺)和两种人工分子(ADM·Na 和 DIM·Na)。我们进行了 DFT 计算以优化它们的分子几何形状,并获得了相应 MD 模拟的原子电子电荷,通过这些我们预测了七种不同的类金刚石和衍生物的自组装结构和模拟轨迹。我们的径向分布函数和结构因子研究表明,这七种类金刚石和衍生物都发生了明显的相转变和自组装。