Tyagi Neetu, Vacek Jonathan C, Givvimani Srikanth, Sen Utpal, Tyagi Suresh C
Department of Physiology and Biophysics, School of Medicine, University of Louisville, Louisville, KY 40292, USA.
J Recept Signal Transduct Res. 2010 Apr;30(2):78-87. doi: 10.3109/10799891003614808.
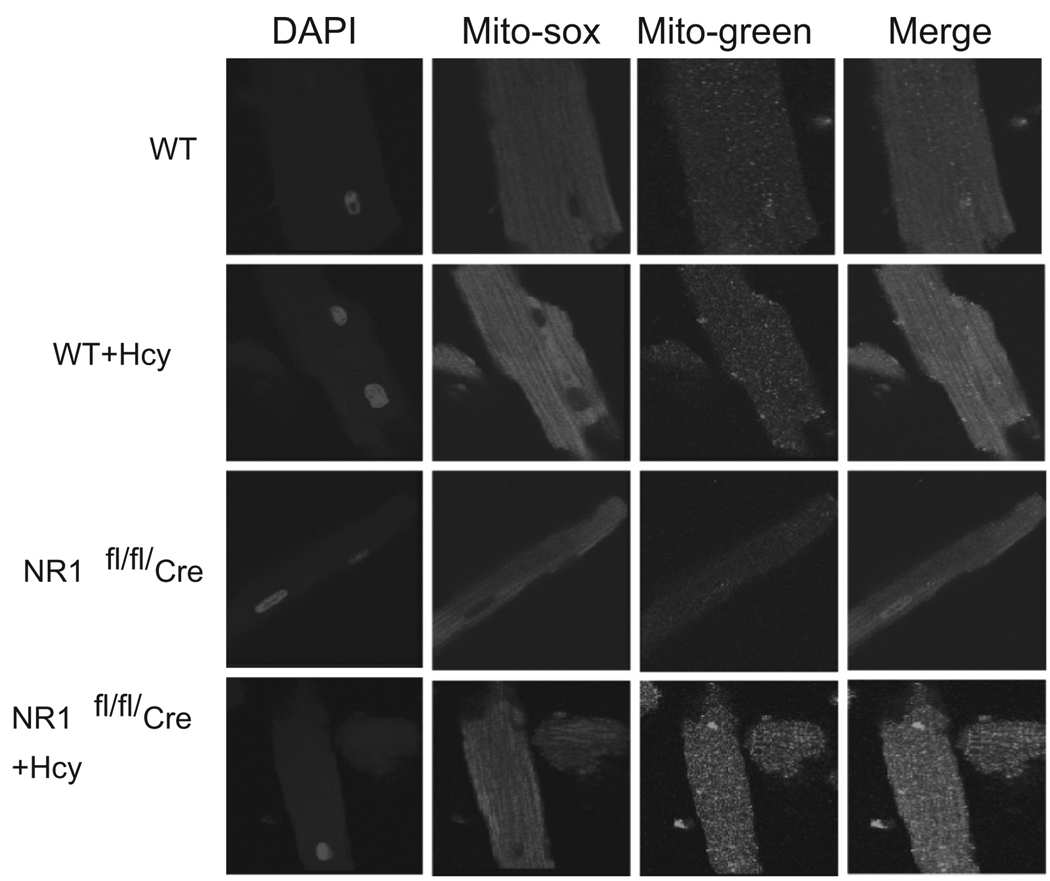
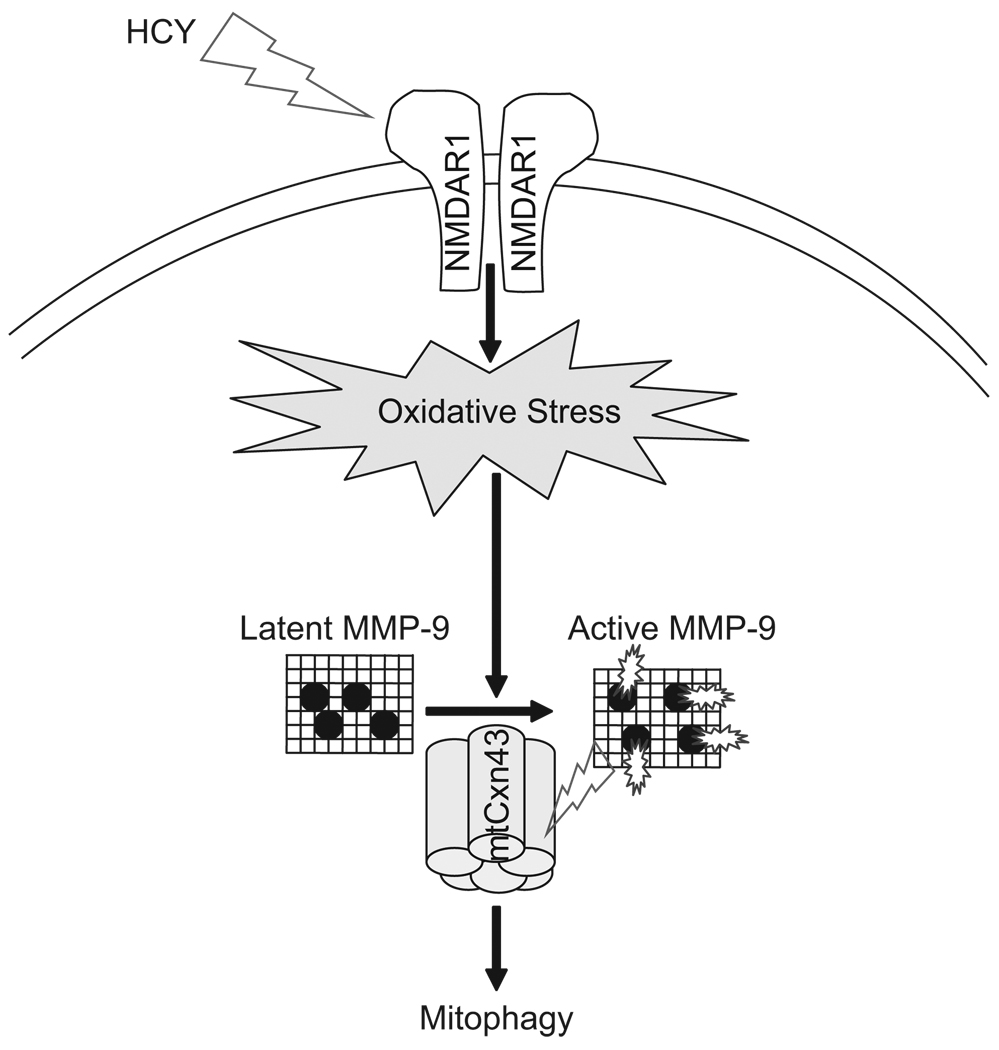
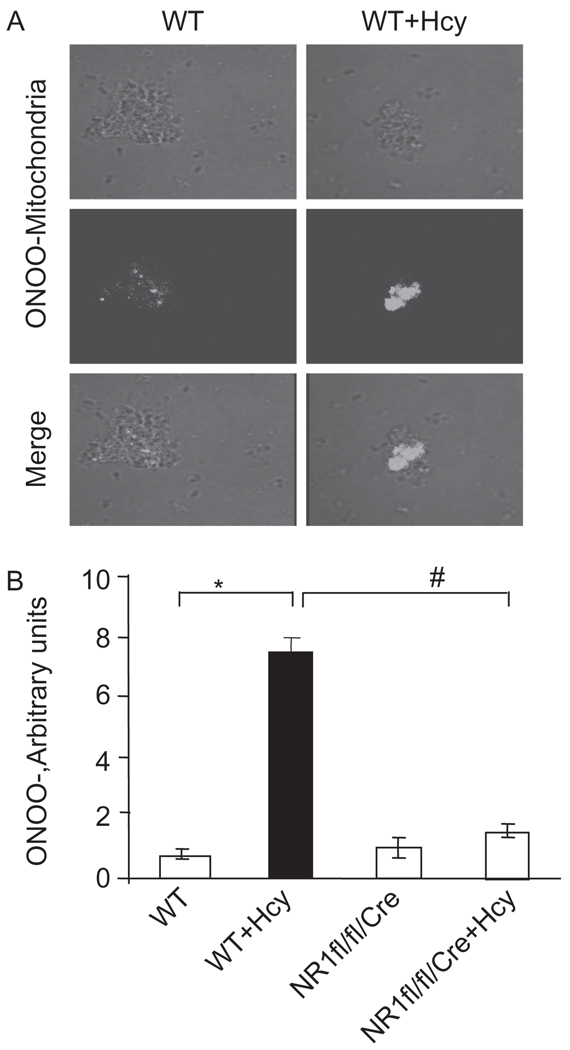
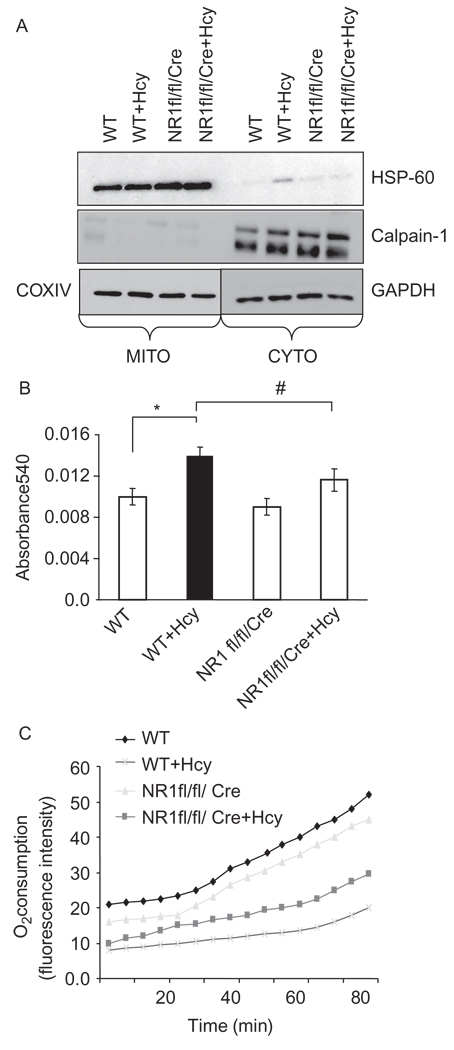
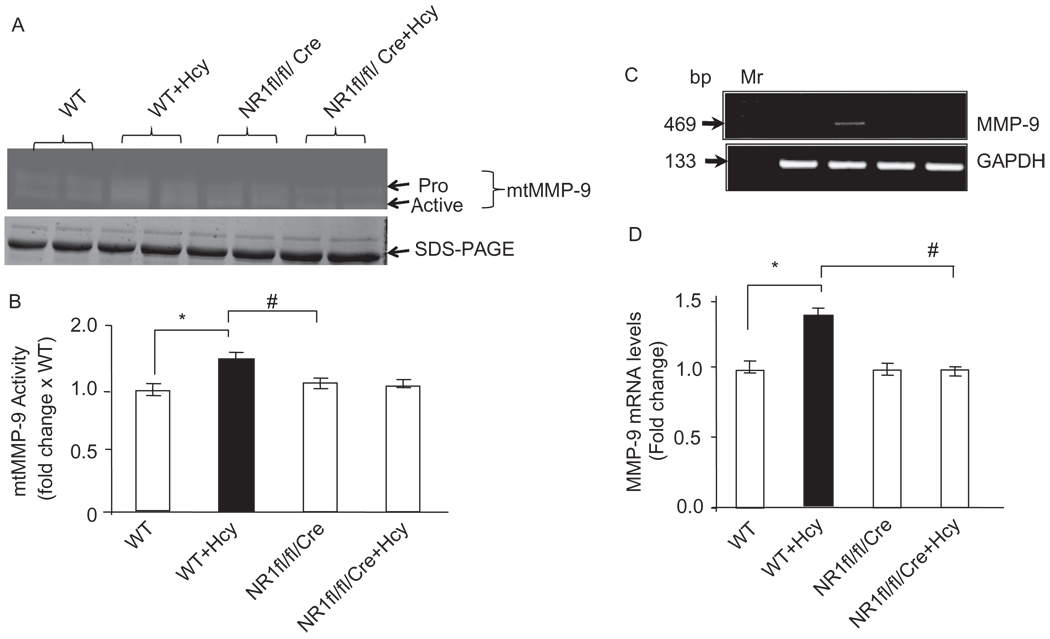
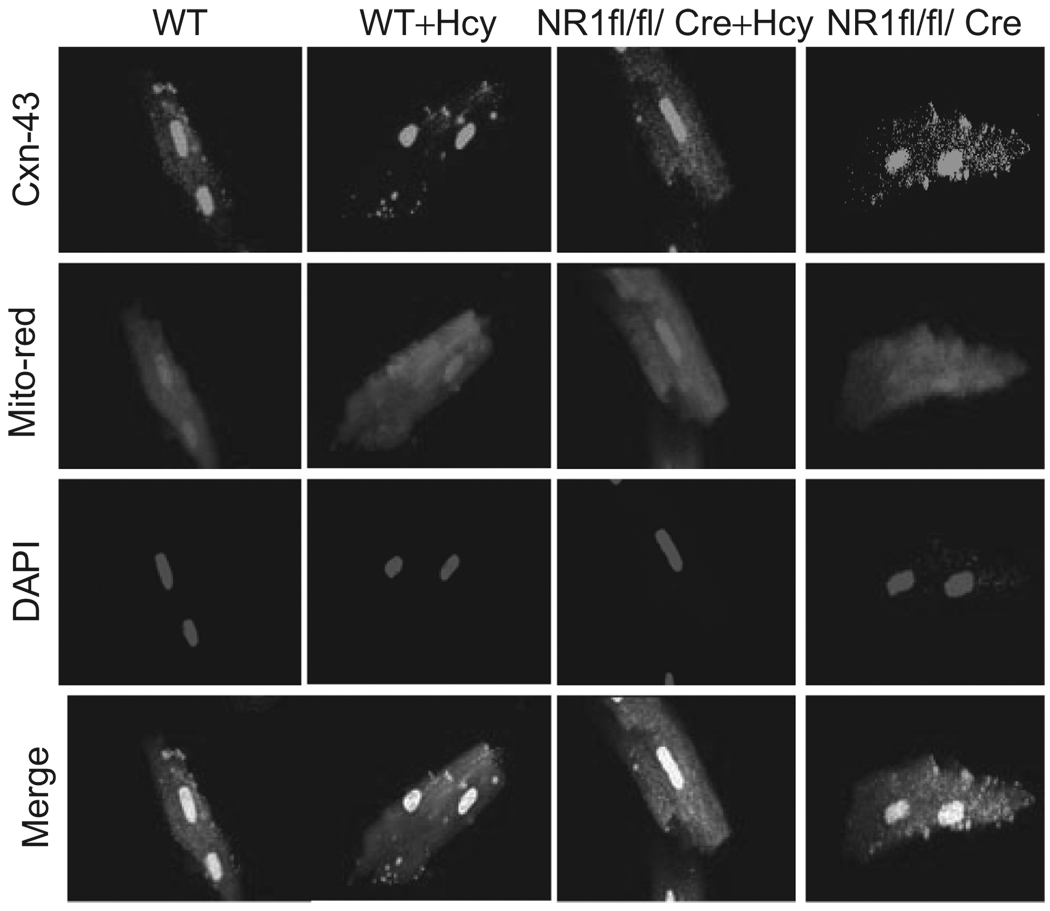
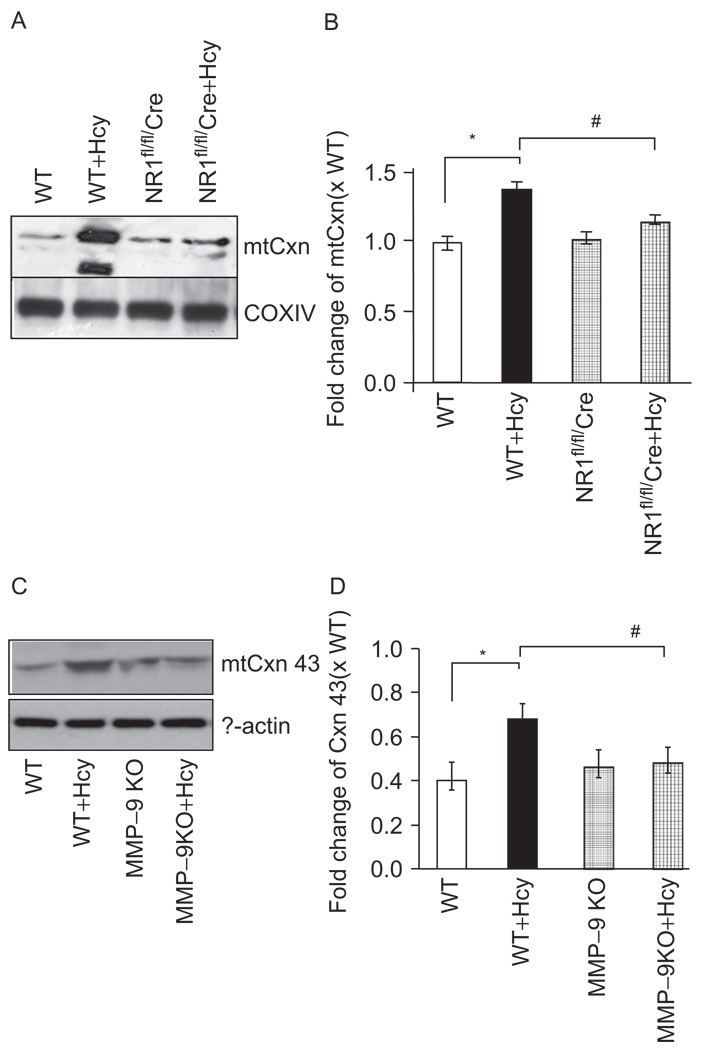
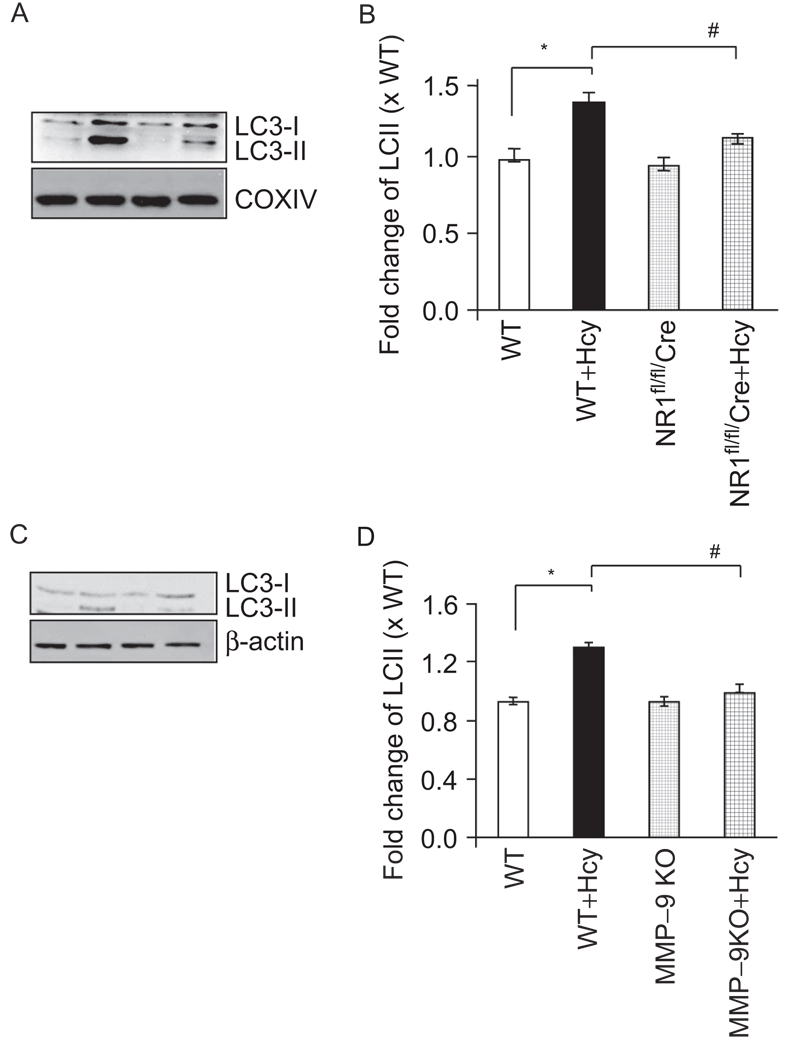
Autophagy is an important process in the pathogenesis of cardiovascular diseases; however, the proximal triggers for mitochondrial autophagy were unknown. The N-methyl-d-aspartate receptor 1 (NMDA-R1) is a receptor for homocysteine (Hcy) and plays a key role in cardiac dysfunction. Cardiac-specific deletion of NMDA-R1 has been shown to ameliorate Hcy-induced myocyte contractility. Hcy activates mitochondrial matrix metalloproteinase-9 (mtMMP-9) and induces translocation of connexin-43 (Cxn-43) to the mitochondria (mtCxn-43). We sought to show cardiac-specific deletion of NMDA-R1 mitigates Hcy-induced mtCxn-43 translocation, mtMMP-9-mediated mtCxn-43 degradation, leading to mitophagy, in part, by decreasing mitochondrial permeability (MPT). Cardiac-specific knockout (KO) of NAMDA-R1 was generated using the cre/lox approach. The myocyte mitochondria were isolated from wild type (WT), WT + Hcy (1.8 g of DL-Hcy/L in the drinking water for 6 weeks), NMDA-R1 KO + Hcy, and NR1(fl/fl)/Cre (NR1(fl/fl)) genetic control mice. Mitochondrial respiratory capacity and MPT were measured by fluorescence-dye methods. The mitochondrial superoxide and peroxinitrite levels were detected by confocal microscopy using Mito-SOX and dihydrorhodamine-123. The mtMMP-9 activity and expression were detected by zymography and RT-PCR analyses. The mtCxn-43 translocation was detected by confocal microscopy. The degradation of mtCxn-43 and LC3-I/II (a marker of autophagy) were detected by Western blot. These results suggested that Hcy enhanced intramitochondrial nitrosative stress in myocytes. There was a robust increase in mtMMP-9 activity. An increase in translocation and degradation of mtCxn-43 was also noted. These increases led to mitophagy. The effects were ameliorated by cardiac-specific deletion of NMDA-R1. We concluded that HHcy increased mitochondrial nitrosative stress, thereby activating mtMMP-9 and inciting the degradation of mtCxn-43. This led to mitophagy, in part, by activating NMDA-R1. The findings of this study will lead to therapeutic ramifications for mitigating cardiovascular diseases by inhibiting the mitochondrial mitophagy and NMDA-R1 receptor.
自噬是心血管疾病发病机制中的一个重要过程;然而,线粒体自噬的近端触发因素尚不清楚。N-甲基-D-天冬氨酸受体1(NMDA-R1)是同型半胱氨酸(Hcy)的受体,在心脏功能障碍中起关键作用。已证明心脏特异性缺失NMDA-R1可改善Hcy诱导的心肌细胞收缩性。Hcy激活线粒体基质金属蛋白酶-9(mtMMP-9)并诱导连接蛋白43(Cxn-43)转位至线粒体(mtCxn-43)。我们试图证明心脏特异性缺失NMDA-R1可减轻Hcy诱导的mtCxn-43转位、mtMMP-9介导的mtCxn-43降解,进而部分通过降低线粒体通透性(MPT)导致线粒体自噬。使用cre/lox方法构建了NMDA-R1的心脏特异性敲除(KO)模型。从野生型(WT)、WT+Hcy(饮用水中含1.8 g DL-Hcy/L,持续6周)、NMDA-R1 KO+Hcy和NR1(fl/fl)/Cre(NR1(fl/fl))基因对照小鼠中分离心肌细胞线粒体。通过荧光染料法测量线粒体呼吸能力和MPT。使用Mito-SOX和二氢罗丹明-123通过共聚焦显微镜检测线粒体超氧化物和过氧亚硝酸盐水平。通过酶谱分析和RT-PCR分析检测mtMMP-9活性和表达。通过共聚焦显微镜检测mtCxn-43转位。通过蛋白质印迹法检测mtCxn-43和LC3-I/II(自噬标志物)的降解。这些结果表明,Hcy增强了心肌细胞线粒体内的亚硝化应激。mtMMP-9活性显著增加。还注意到mtCxn-43的转位和降解增加。这些增加导致线粒体自噬。心脏特异性缺失NMDA-R1可改善这些效应。我们得出结论,高Hcy血症增加了线粒体亚硝化应激,从而激活mtMMP-9并引发mtCxn-43的降解。这部分通过激活NMDA-R1导致线粒体自噬。本研究结果将为通过抑制线粒体自噬和NMDA-R1受体减轻心血管疾病带来治疗意义。