Division of Mathematical Biology, MRC National Institute for Medical Research, London, UK.
BMC Bioinformatics. 2010 Feb 20;11:97. doi: 10.1186/1471-2105-11-97.
The hierarchical and partially redundant nature of protein structures justifies the definition of frequently occurring conformations of short fragments as 'states'. Collections of selected representatives for these states define Structural Alphabets, describing the most typical local conformations within protein structures. These alphabets form a bridge between the string-oriented methods of sequence analysis and the coordinate-oriented methods of protein structure analysis.
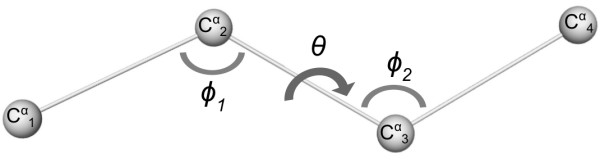
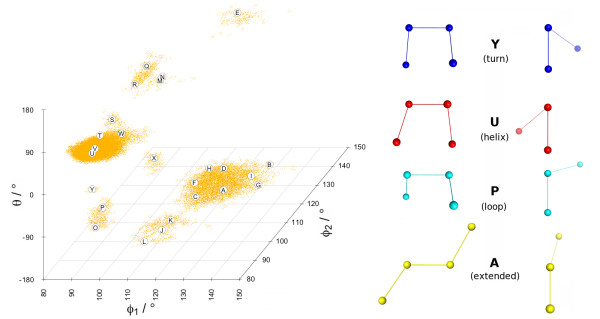
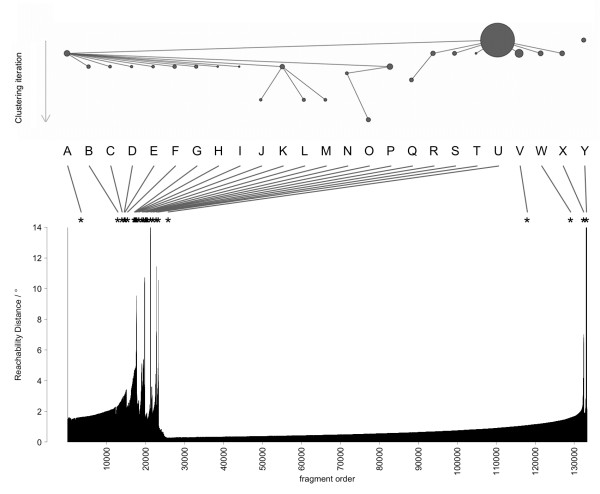
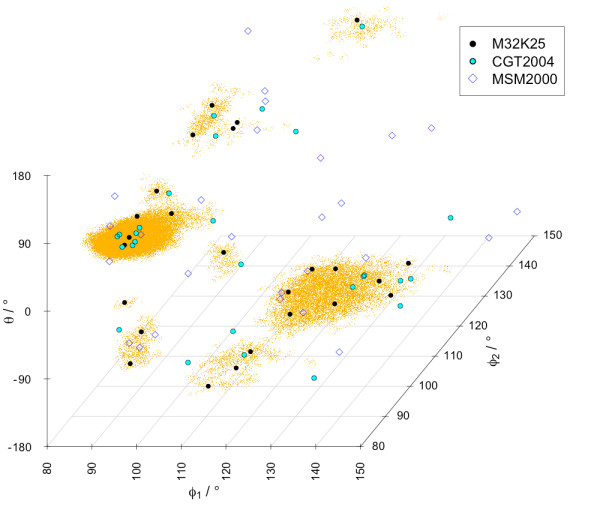
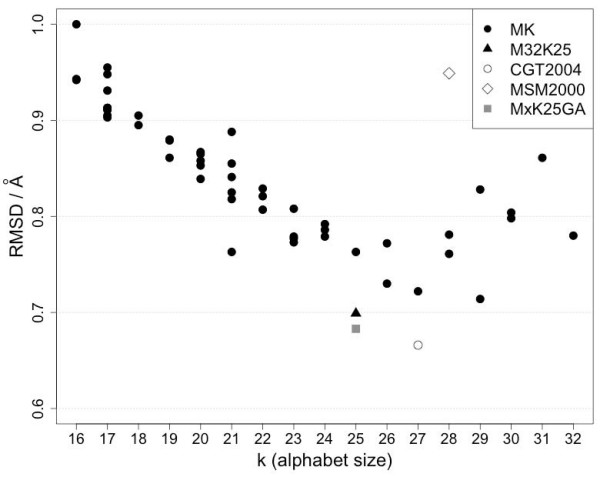
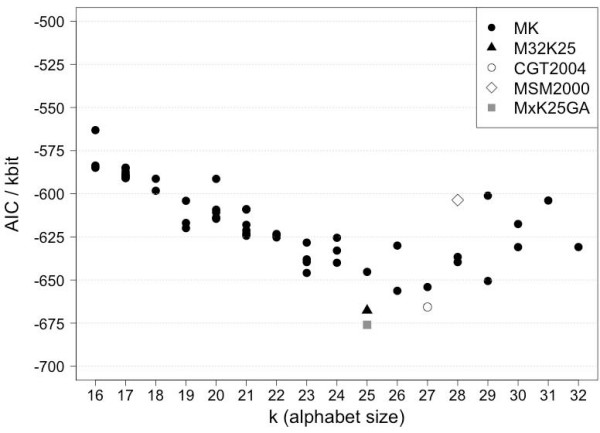
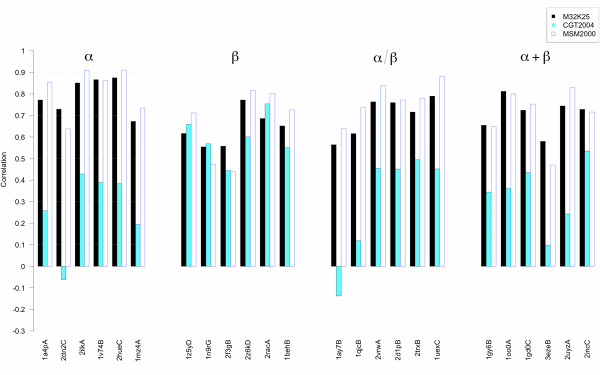
A Structural Alphabet has been derived by clustering all four-residue fragments of a high-resolution subset of the protein data bank and extracting the high-density states as representative conformational states. Each fragment is uniquely defined by a set of three independent angles corresponding to its degrees of freedom, capturing in simple and intuitive terms the properties of the conformational space. The fragments of the Structural Alphabet are equivalent to the conformational attractors and therefore yield a most informative encoding of proteins. Proteins can be reconstructed within the experimental uncertainty in structure determination and ensembles of structures can be encoded with accuracy and robustness.
The density-based Structural Alphabet provides a novel tool to describe local conformations and it is specifically suitable for application in studies of protein dynamics.
蛋白质结构的层次和部分冗余性质证明了将短片段的频繁出现构象定义为“状态”是合理的。这些状态的选定代表集合定义了结构字母表,描述了蛋白质结构中最典型的局部构象。这些字母表在序列分析的面向字符串的方法和蛋白质结构分析的面向坐标的方法之间架起了桥梁。
通过对蛋白质数据库的高分辨率子集的所有四残基片段进行聚类,并提取高密度状态作为代表性构象状态,得出了一个结构字母表。每个片段都由一组对应于其自由度的三个独立角度唯一定义,用简单直观的术语捕捉构象空间的性质。结构字母表的片段等同于构象吸引子,因此提供了对蛋白质的最有信息的编码。可以在结构确定的实验不确定性内重建蛋白质,并且可以准确和稳健地对结构集合进行编码。
基于密度的结构字母表提供了一种描述局部构象的新工具,特别适合于蛋白质动力学研究。