Department of Biological Sciences, National University of Singapore, Singapore, Singapore.
PLoS One. 2010 Feb 18;5(2):e8943. doi: 10.1371/journal.pone.0008943.
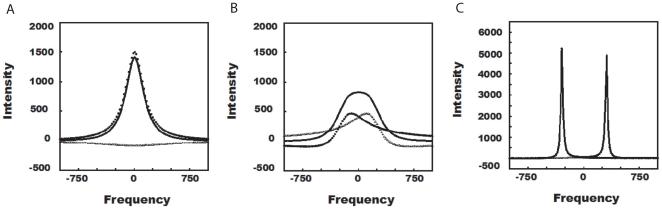
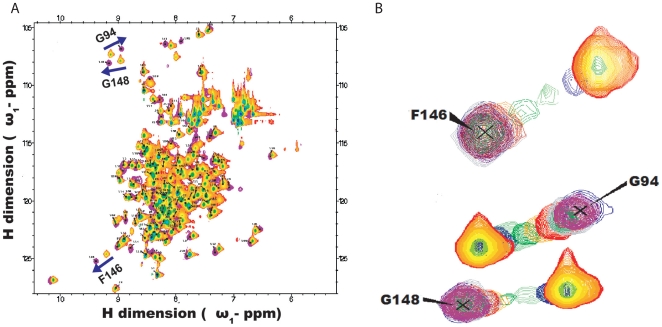
Nuclear Magnetic Resonance (NMR) spectroscopy offers a variety of experiments to study protein-ligand interactions at atomic resolution. Among these experiments, 15N Heteronuclear Single Quantum Correlation (HSQC)experiment is simple, less time consuming and highly informative in mapping the binding site of the ligand. The interpretation of 15N HSQC becomes ambiguous when the chemical shift perturbations are caused by non-specific interactions like allosteric changes and local structural rearrangement. Under such cases, detailed chemical exchange analysis based on chemical shift perturbation will assist in locating the binding site accurately.
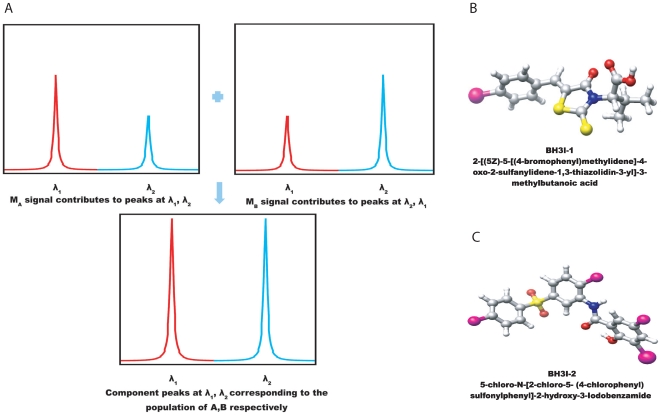
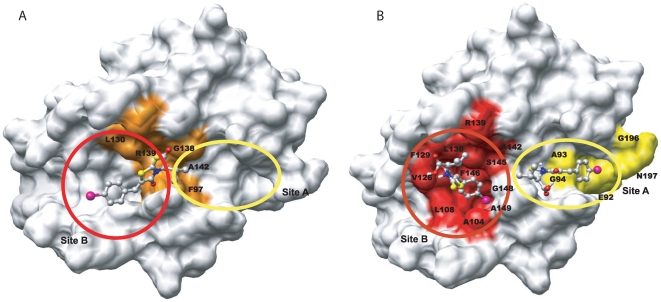
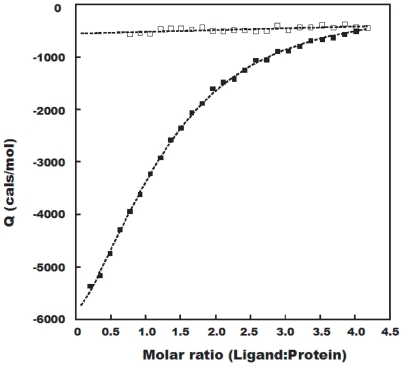
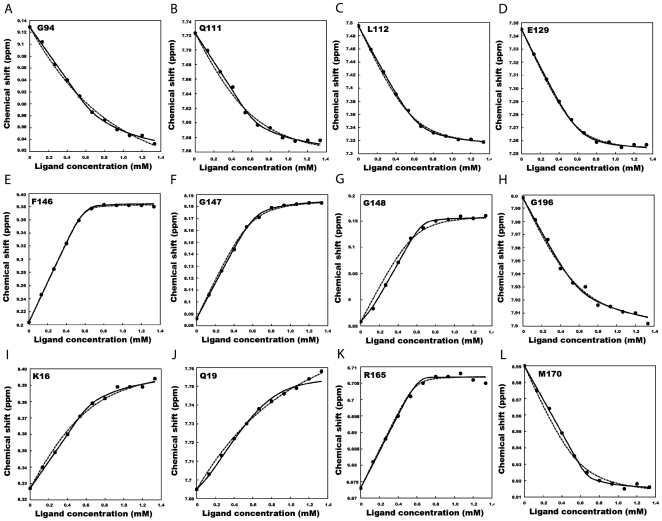
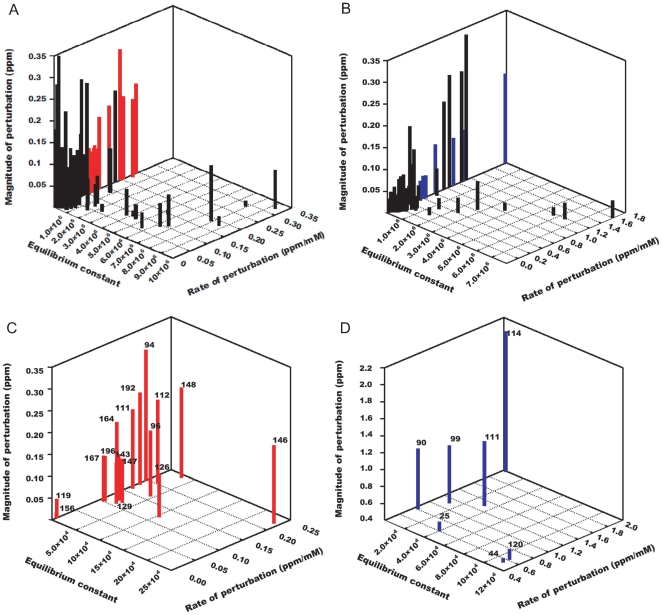
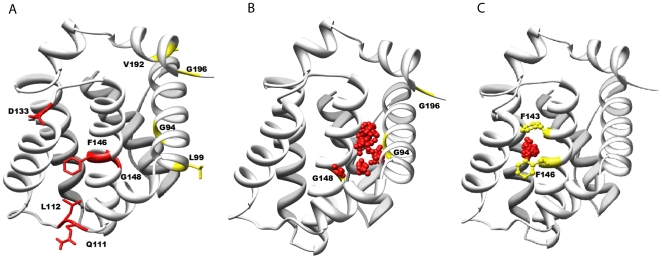
METHODOLOGY/PRINCIPAL FINDINGS: We have automated the mapping of binding sites for fast chemical exchange systems using information obtained from 15N HSQC spectra of protein serially titrated with ligand of increasing concentrations. The automated program Auto-FACE (Auto-FAst Chemical Exchange analyzer) determines the parameters, e.g. rate of change of perturbation, binding equilibrium constant and magnitude of chemical shift perturbation to map the binding site residues.Interestingly, the rate of change of perturbation at lower ligand concentration is highly sensitive in differentiating the binding site residues from the non-binding site residues. To validate this program, the interaction between the protein hBcl(XL) and the ligand BH3I-1 was studied. Residues in the hydrophobic BH3 binding groove of hBcl(XL) were easily identified to be crucial for interaction with BH3I-1 from other residues that also exhibited perturbation. The geometrically averaged equilibrium constant (3.0 x 10(4)) calculated for the residues present at the identified binding site is consistent with the values obtained by other techniques like isothermal calorimetry and fluorescence polarization assays (12.8 x 10(4)). Adjacent to the primary site, an additional binding site was identified which had an affinity of 3.8 times weaker than the former one. Further NMR based model fitting for individual residues suggest single site model for residues present at these binding sites and two site model for residues present between these sites. This implies that chemical shift perturbation can represent the local binding event much more accurately than the global binding event.
CONCLUSION/SIGNIFICANCE: Detail NMR chemical shift perturbation analysis enabled binding site residues to be distinguished from non-binding site residues for accurate mapping of interaction site in complex fast exchange system between small molecule and protein. The methodology is automated and implemented in a program called "Auto-FACE", which also allowed quantitative information of each interaction site and elucidation of binding mechanism.
核磁共振(NMR)光谱提供了多种实验方法,可在原子分辨率下研究蛋白质-配体相互作用。在这些实验中,15N 异核单量子相关(HSQC)实验简单、耗时少且信息量丰富,可用于绘制配体的结合位点。当化学位移扰动是由变构变化和局部结构重排等非特异性相互作用引起时,15N HSQC 的解释就变得不明确。在这种情况下,基于化学位移扰动的详细化学交换分析将有助于准确确定结合位点。
方法/主要发现:我们使用从蛋白质与配体连续滴定的 15N HSQC 光谱中获得的信息,自动化了快速化学交换系统结合位点的映射。自动程序 Auto-FACE(Auto-FAst Chemical Exchange analyzer)确定了参数,例如扰动变化率、结合平衡常数和化学位移扰动的幅度,以绘制结合位点残基。有趣的是,较低配体浓度下的扰动变化率在区分结合位点残基和非结合位点残基方面非常敏感。为了验证该程序,研究了蛋白 hBcl(XL)与配体 BH3I-1 的相互作用。hBcl(XL) 的疏水性 BH3 结合槽中的残基很容易被鉴定为与 BH3I-1 相互作用的关键残基,而其他也表现出扰动的残基则不是。从其他技术(如等温量热法和荧光偏振测定法)获得的与鉴定结合位点上的残基相对应的几何平均平衡常数(3.0×10(4))是一致的。在主要结合位点旁边,还鉴定到了一个额外的结合位点,其亲和力比前者弱 3.8 倍。对各个残基的进一步 NMR 模型拟合表明,这些结合位点上的残基采用单一位点模型,而这些位点之间的残基采用双一位点模型。这意味着化学位移扰动可以比全局结合事件更准确地代表局部结合事件。
结论/意义:详细的 NMR 化学位移扰动分析使结合位点残基能够与非结合位点残基区分开来,从而在小分子与蛋白质之间的复杂快速交换系统中准确绘制相互作用位点。该方法已自动化,并实现为一个名为“Auto-FACE”的程序,该程序还允许获得每个相互作用位点的定量信息,并阐明结合机制。