Mines ParisTech, Centre for Computational Biology, Fontainbleau, France.
BMC Bioinformatics. 2010 Feb 22;11:99. doi: 10.1186/1471-2105-11-99.
Predicting which molecules can bind to a given binding site of a protein with known 3D structure is important to decipher the protein function, and useful in drug design. A classical assumption in structural biology is that proteins with similar 3D structures have related molecular functions, and therefore may bind similar ligands. However, proteins that do not display any overall sequence or structure similarity may also bind similar ligands if they contain similar binding sites. Quantitatively assessing the similarity between binding sites may therefore be useful to propose new ligands for a given pocket, based on those known for similar pockets.
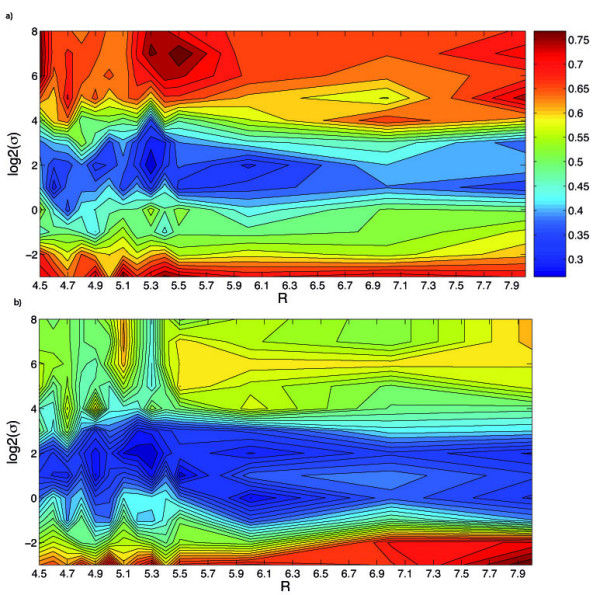
We propose a new method to quantify the similarity between binding pockets, and explore its relevance for ligand prediction. We represent each pocket by a cloud of atoms, and assess the similarity between two pockets by aligning their atoms in the 3D space and comparing the resulting configurations with a convolution kernel. Pocket alignment and comparison is possible even when the corresponding proteins share no sequence or overall structure similarities. In order to predict ligands for a given target pocket, we compare it to an ensemble of pockets with known ligands to identify the most similar pockets. We discuss two criteria to evaluate the performance of a binding pocket similarity measure in the context of ligand prediction, namely, area under ROC curve (AUC scores) and classification based scores. We show that the latter is better suited to evaluate the methods with respect to ligand prediction, and demonstrate the relevance of our new binding site similarity compared to existing similarity measures.
This study demonstrates the relevance of the proposed method to identify ligands binding to known binding pockets. We also provide a new benchmark for future work in this field. The new method and the benchmark are available at http://cbio.ensmp.fr/paris/.
预测具有已知 3D 结构的蛋白质的特定结合位点可以结合哪些分子,对于破译蛋白质功能非常重要,并且在药物设计中也很有用。结构生物学中的一个经典假设是,具有相似 3D 结构的蛋白质具有相关的分子功能,因此可能结合相似的配体。然而,如果蛋白质即使不显示任何整体序列或结构相似性,如果它们包含相似的结合位点,也可能结合相似的配体。因此,定量评估结合位点之间的相似性可能有助于根据已知的相似口袋提出给定口袋的新配体。
我们提出了一种新的方法来量化结合口袋之间的相似性,并探讨了其对配体预测的相关性。我们通过原子云表示每个口袋,并通过在 3D 空间中对齐它们的原子,然后将得到的构象与卷积核进行比较,来评估两个口袋之间的相似性。即使对应的蛋白质没有共享序列或整体结构相似性,口袋对齐和比较也是可能的。为了预测给定靶口袋的配体,我们将其与具有已知配体的口袋集合进行比较,以识别最相似的口袋。我们讨论了两种评估结合口袋相似性度量在配体预测背景下的性能的标准,即 ROC 曲线下的面积(AUC 分数)和基于分类的分数。我们表明,后者更适合于根据配体预测评估方法,并展示了与现有相似性度量相比,我们新的结合位点相似性的相关性。
本研究证明了所提出的方法对于识别已知结合口袋的配体的相关性。我们还提供了该领域未来工作的新基准。新方法和基准可在 http://cbio.ensmp.fr/paris/ 获得。