Department of Neurology and Department of Human Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA.
Brain. 2010 Mar;133(Pt 3):671-89. doi: 10.1093/brain/awq013. Epub 2010 Feb 22.
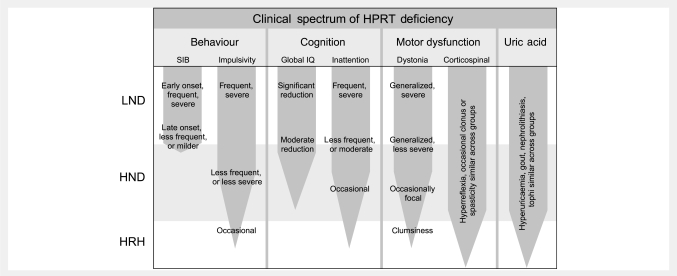
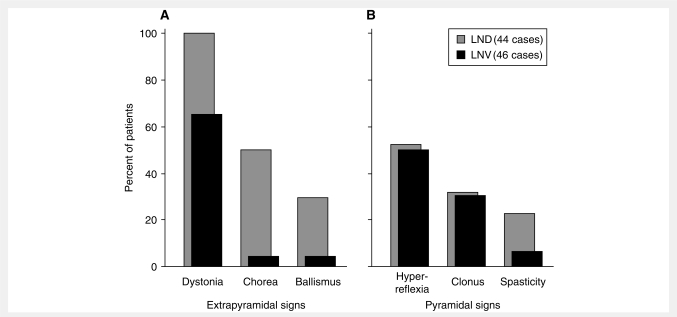
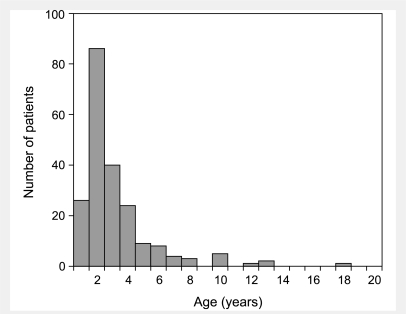
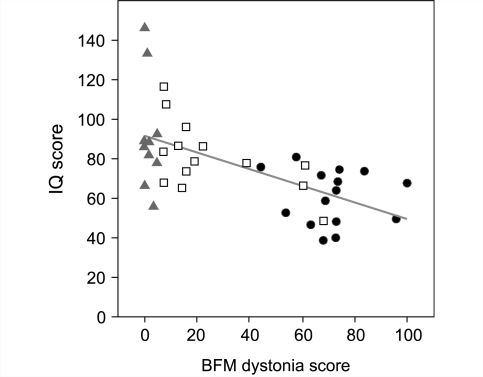
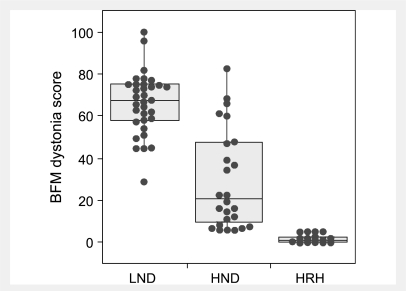
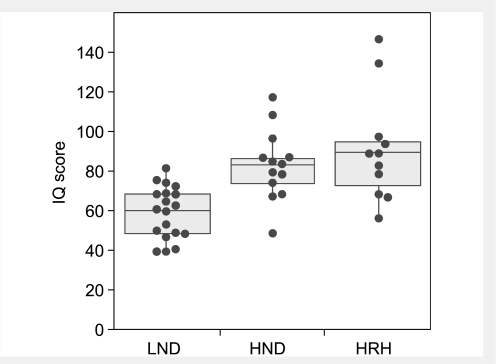
Lesch-Nyhan disease is a neurogenetic disorder caused by deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase. The classic form of the disease is described by a characteristic syndrome that includes overproduction of uric acid, severe generalized dystonia, cognitive disability and self-injurious behaviour. In addition to the classic disease, variant forms of the disease occur wherein some clinical features are absent or unusually mild. The current studies provide the results of a prospective and multi-centre international study focusing on neurological manifestations of the largest cohort of Lesch-Nyhan disease variants evaluated to date, with 46 patients from 3 to 65 years of age coming from 34 families. All had evidence for overproduction of uric acid. Motor abnormalities were evident in 42 (91%), ranging from subtle clumsiness to severely disabling generalized dystonia. Cognitive function was affected in 31 (67%) but it was never severe. Though none exhibited self-injurious behaviours, many exhibited behaviours that were maladaptive. Only three patients had no evidence of neurological dysfunction. Our results were compared with a comprehensive review of 78 prior reports describing a total of 127 Lesch-Nyhan disease variants. Together these results define the spectrum of clinical features associated with hypoxanthine-guanine phosphoribosyltransferase deficiency. At one end of the spectrum are patients with classic Lesch-Nyhan disease and the full clinical phenotype. At the other end of the spectrum are patients with overproduction of uric acid but no apparent neurological or behavioural deficits. Inbetween are patients with varying degrees of motor, cognitive, or behavioural abnormalities. Recognition of this spectrum is valuable for understanding the pathogenesis and diagnosis of all forms of hypoxanthine-guanine phosphoribosyltransferase deficiency.
莱施-尼汉病是一种神经遗传疾病,由次黄嘌呤-鸟嘌呤磷酸核糖转移酶缺乏引起。经典型疾病表现为一种特征性综合征,包括尿酸生成过多、严重的全身性肌张力障碍、认知障碍和自伤行为。除了经典疾病外,还存在变体形式的疾病,其中一些临床特征缺失或异常轻微。目前的研究提供了一项前瞻性、多中心国际研究的结果,该研究聚焦于迄今为止评估的最大队列的莱施-尼汉病变体的神经表现,共有 34 个家庭的 46 名 3 至 65 岁的患者参与。所有患者均有尿酸生成过多的证据。42 名(91%)患者存在运动异常,从轻微笨拙到严重致残性全身性肌张力障碍不等。31 名(67%)患者认知功能受损,但从未严重受损。虽然没有患者表现出自伤行为,但许多患者表现出适应不良的行为。只有 3 名患者没有神经功能障碍的证据。我们的结果与对描述总共 127 种莱施-尼汉病变体的 78 份先前报告的全面综述进行了比较。这些结果共同定义了与次黄嘌呤-鸟嘌呤磷酸核糖转移酶缺乏相关的临床特征谱。一端是具有经典莱施-尼汉病和完整临床表型的患者。另一端是尿酸生成过多但没有明显神经或行为缺陷的患者。在两者之间是存在不同程度运动、认知或行为异常的患者。认识到这种谱对于理解所有形式的次黄嘌呤-鸟嘌呤磷酸核糖转移酶缺乏症的发病机制和诊断都很有价值。