Department of Biochemistry and Molecular Cell Biology, University Hospital, RWTH Aachen University, Aachen, Germany.
J Cell Mol Med. 2011 Mar;15(3):668-78. doi: 10.1111/j.1582-4934.2010.01041.x.
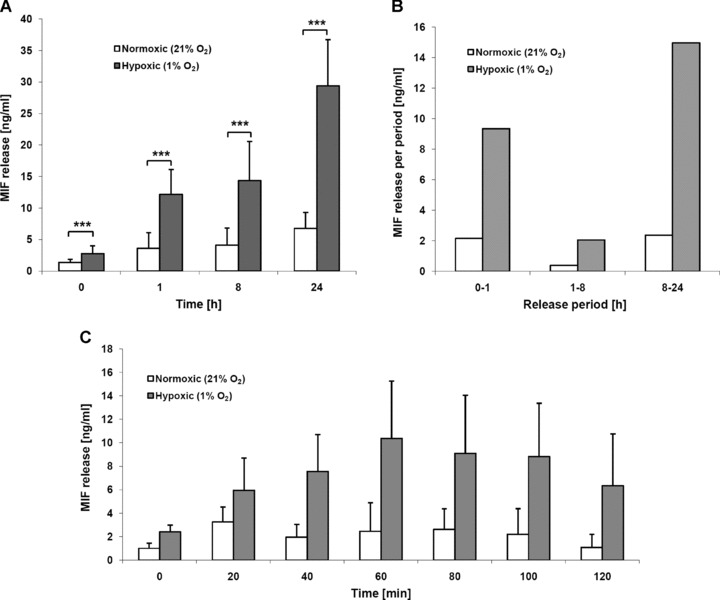
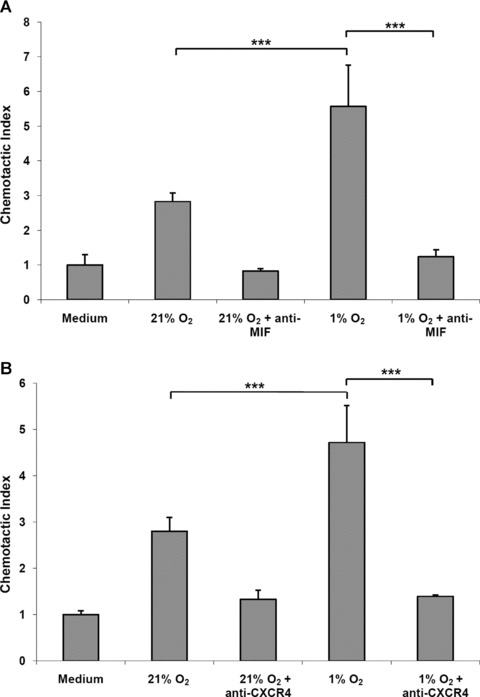
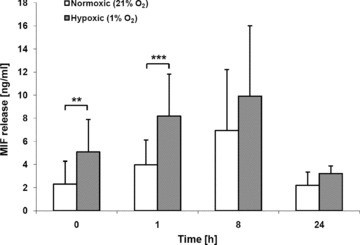
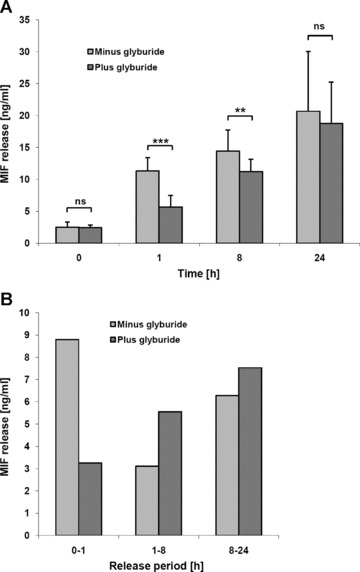
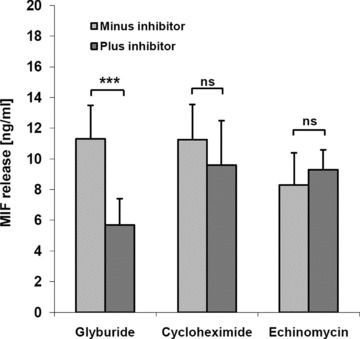
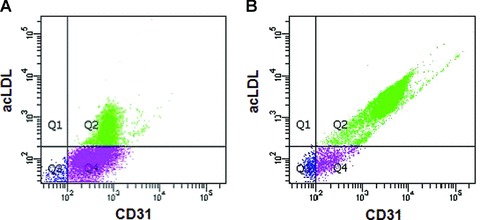
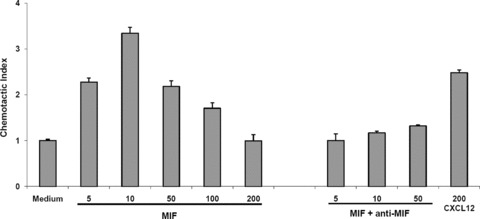
Macrophage migration inhibitory factor (MIF) is a pleiotropic inflammatory cytokine that was recently identified as a non-cognate ligand of the CXC-family chemokine receptors 2 and 4 (CXCR2 and CXCR4). MIF is expressed and secreted from endothelial cells (ECs) following atherogenic stimulation, exhibits chemokine-like properties and promotes the recruitment of leucocytes to atherogenic endothelium. CXCR4 expressed on endothelial progenitor cells (EPCs) and EC-derived CXCL12, the cognate ligand of CXCR4, have been demonstrated to be critical when EPCs are recruited to ischemic tissues. Here we studied whether hypoxic stimulation triggers MIF secretion from ECs and whether the MIF/CXCR4 axis contributes to EPC recruitment. Exposure of human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAoECs) to 1% hypoxia led to the specific release of substantial amounts of MIF. Hypoxia-induced MIF release followed a biphasic behaviour. MIF secretion in the first phase peaked at 60 min. and was inhibited by glyburide, indicating that this MIF pool was secreted by a non-classical mechanism and originated from pre-formed MIF stores. Early hypoxia-triggered MIF secretion was not inhibited by cycloheximide and echinomycin, inhibitors of general and hypoxia-inducible factor (HIF)-1α-induced protein synthesis, respectively. A second phase of MIF secretion peaked around 8 hrs and was likely due to HIF-1α-induced de novo synthesis of MIF. To functionally investigate the role of hypoxia-inducible secreted MIF on the recruitment of EPCs, we subjected human AcLDL(+) KDR(+) CD31(+) EPCs to a chemotactic MIF gradient. MIF potently promoted EPC chemotaxis in a dose-dependent bell-shaped manner (peak: 10 ng/ml MIF). Importantly, EPC migration was induced by supernatants of hypoxia-conditioned HUVECs, an effect that was completely abrogated by anti-MIF- or anti-CXCR4-antibodies. Thus, hypoxia-induced MIF secretion from ECs might play an important role in the recruitment and migration of EPCs to hypoxic tissues such as after ischemia-induced myocardial damage.
巨噬细胞移动抑制因子(MIF)是一种多效性炎症细胞因子,最近被鉴定为 CXC 家族趋化因子受体 2 和 4(CXCR2 和 CXCR4)的非配体。MIF 在动脉粥样硬化刺激后从内皮细胞(ECs)表达和分泌,表现出趋化因子样特性,并促进白细胞向动脉粥样硬化内皮的募集。在内皮祖细胞(EPCs)上表达的 CXCR4 和 EC 衍生的 CXCL12(CXCR4 的配体),已被证明在 EPC 被招募到缺血组织时至关重要。在这里,我们研究了缺氧刺激是否会触发 EC 释放 MIF,以及 MIF/CXCR4 轴是否有助于 EPC 的募集。将人脐静脉内皮细胞(HUVECs)和人主动脉内皮细胞(HAoECs)暴露于 1%缺氧会导致大量 MIF 的特异性释放。缺氧诱导的 MIF 释放呈双相行为。第一阶段的 MIF 分泌在 60 分钟达到峰值,并被格列本脲抑制,表明该 MIF 池是通过非经典机制释放的,并且来源于预先形成的 MIF 储存库。早期缺氧触发的 MIF 分泌不受环己酰亚胺和依诺霉素的抑制,环己酰亚胺和依诺霉素分别是一般和缺氧诱导因子(HIF)-1α诱导的蛋白质合成抑制剂。MIF 分泌的第二阶段在大约 8 小时达到峰值,可能是由于 HIF-1α诱导的 MIF 从头合成。为了功能研究缺氧诱导的分泌型 MIF 在 EPC 募集中的作用,我们使人类 AcLDL(+)KDR(+)CD31(+)EPC 接受趋化性 MIF 梯度。MIF 以剂量依赖性钟形方式强烈促进 EPC 趋化(峰值:10ng/ml MIF)。重要的是,缺氧条件下 HUVEC 的上清液诱导 EPC 迁移,该作用可被抗 MIF 或抗 CXCR4 抗体完全阻断。因此,EC 中缺氧诱导的 MIF 分泌可能在 EPC 向缺氧组织(如缺血性心肌损伤后)的募集和迁移中发挥重要作用。