Mayo Clinic, Rochester, MN 55905, USA.
Adv Chronic Kidney Dis. 2010 Mar;17(2):131-9. doi: 10.1053/j.ackd.2009.12.004.
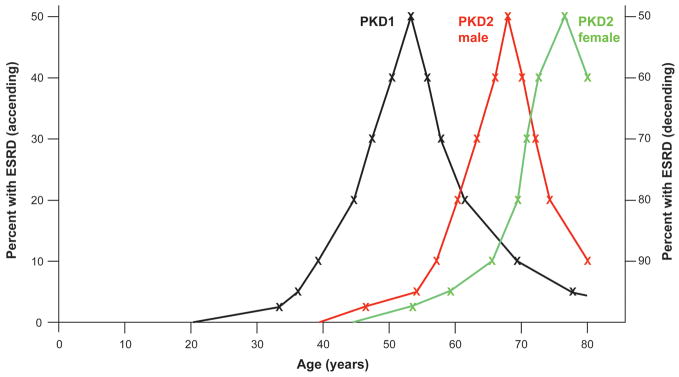
In common with other Mendelian diseases, the presentation and progression of autosomal dominant polycystic kidney disease (ADPKD) vary widely in the population. The typical course is of adult-onset disease with ESRD in the 6th decade. However, a small proportion has adequate renal function into the 9th decade, whereas others present with enlarged kidneys as neonates. ADPKD is genetically heterogeneous, and the disease gene is a major determinant of severity; PKD1 on average is associated with ESRD 20 years earlier than PKD2. The majority of PKD1 and PKD2 mutations are likely fully inactivating although recent studies indicate that some alleles retain partial activity (hypomorphic alleles). Homozygotes for such alleles are viable and in combination with an inactivating allele can result in early-onset disease. Hypomorphic alleles and mosaicism may also account for some cases with unusually mild disease. The degree of phenotypic variation detected in families indicates that genetic background influences disease severity. Genome-wide association studies are planned to map common variants associated with severity. Although ADPKD is a simple genetic disease, fully understanding the phenotypic variability requires consideration of influences at the genic, allelic, and genetic background level, and so, ultimately, it is complex.
与其他孟德尔疾病一样,常染色体显性多囊肾病 (ADPKD) 在人群中的表现和进展差异很大。典型的病程是成年起病,60 岁时出现终末期肾病。然而,一小部分人到 90 岁时仍有足够的肾功能,而另一些人则在新生儿期就出现肾脏增大。ADPKD 具有遗传异质性,疾病基因是严重程度的主要决定因素;PKD1 平均比 PKD2 早 20 年出现终末期肾病。尽管最近的研究表明,一些等位基因保留部分活性(低功能等位基因),但大多数 PKD1 和 PKD2 突变可能完全失活。这些等位基因的纯合子是有活力的,与失活等位基因结合可导致早发性疾病。低功能等位基因和嵌合性也可能导致一些疾病表现异常轻微的病例。在家族中检测到的表型变异程度表明遗传背景影响疾病严重程度。计划进行全基因组关联研究以绘制与严重程度相关的常见变异。尽管 ADPKD 是一种简单的遗传疾病,但要充分了解表型的可变性,需要考虑基因、等位基因和遗传背景水平的影响,因此,最终它是复杂的。