Bioinformatics Centre, Institute of Microbial Technology, Sector-39A, Chandigarh, India.
BMC Bioinformatics. 2010 Mar 11;11:125. doi: 10.1186/1471-2105-11-125.
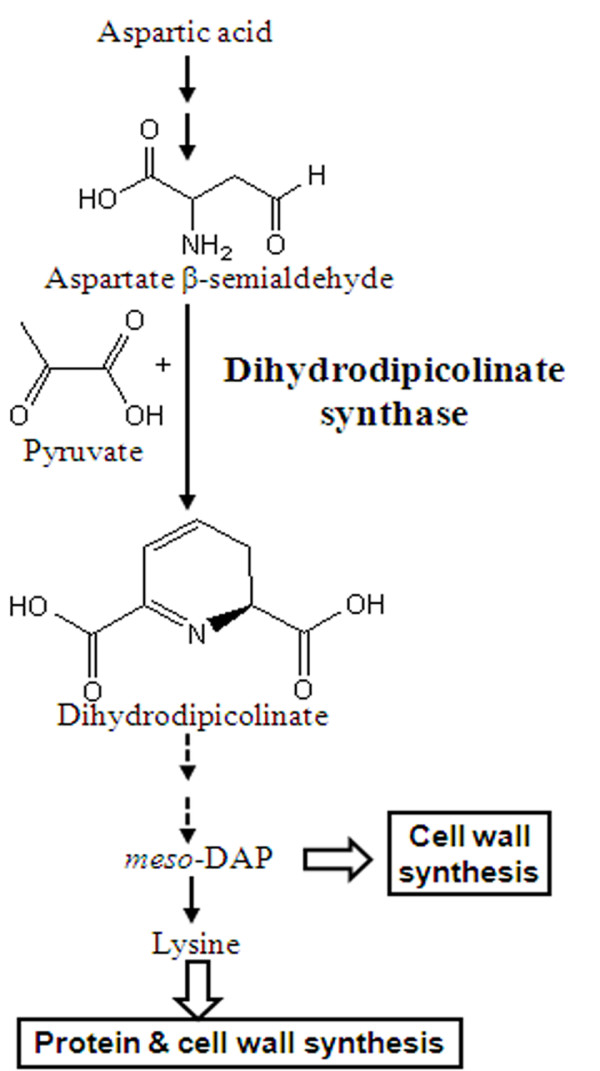
Identification of novel drug targets and their inhibitors is a major challenge in the field of drug designing and development. Diaminopimelic acid (DAP) pathway is a unique lysine biosynthetic pathway present in bacteria, however absent in mammals. This pathway is vital for bacteria due to its critical role in cell wall biosynthesis. One of the essential enzymes of this pathway is dihydrodipicolinate synthase (DHDPS), considered to be crucial for the bacterial survival. In view of its importance, the development and prediction of potent inhibitors against DHDPS may be valuable to design effective drugs against bacteria, in general.
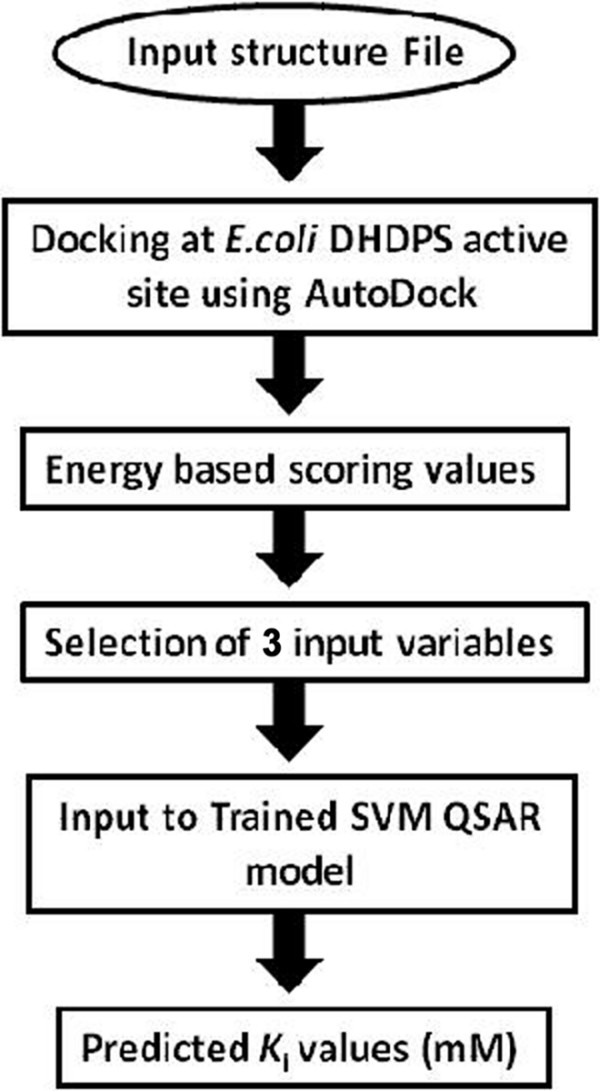
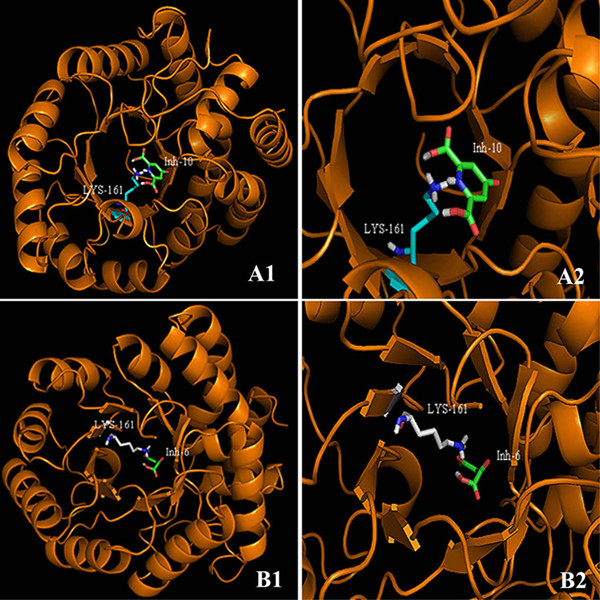
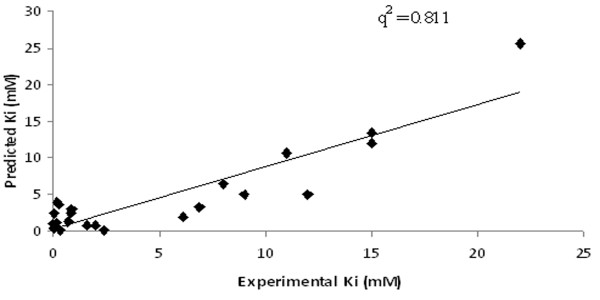
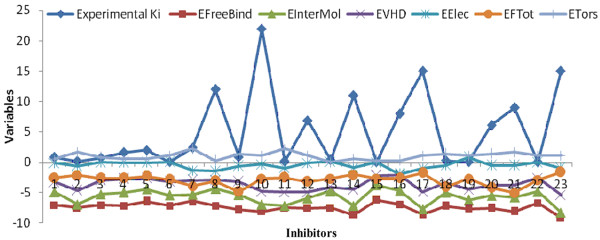
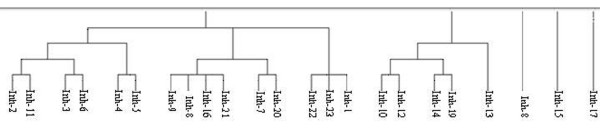
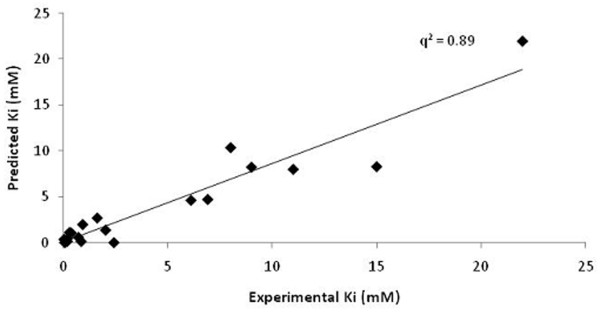
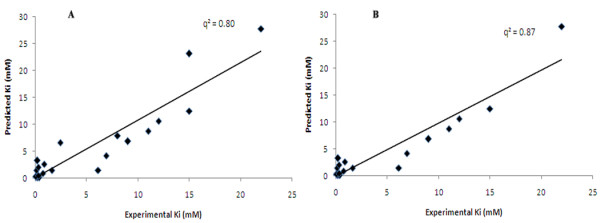
This paper describes a methodology for predicting novel/potent inhibitors against DHDPS. Here, quantitative structure activity relationship (QSAR) models were trained and tested on experimentally verified 23 enzyme's inhibitors having inhibitory value (Ki) in the range of 0.005-22(mM). These inhibitors were docked at the active site of DHDPS (1YXD) using AutoDock software, which resulted in 11 energy-based descriptors. For QSAR modeling, Multiple Linear Regression (MLR) model was engendered using best four energy-based descriptors yielding correlation values R/q2 of 0.82/0.67 and MAE of 2.43. Additionally, Support Vector Machine (SVM) based model was developed with three crucial descriptors selected using F-stepping remove-one approach, which enhanced the performance by attaining R/q2 values of 0.93/0.80 and MAE of 1.89. To validate the performance of QSAR models, external cross-validation procedure was adopted which accomplished high training/testing correlation values (q2/r2) in the range of 0.78-0.83/0.93-0.95.
Our results suggests that ligand-receptor binding interactions for DHDPS employing QSAR modeling seems to be a promising approach for prediction of antibacterial agents. To serve the experimentalist to develop novel/potent inhibitors, a webserver "KiDoQ" has been developed http://crdd.osdd.net/raghava/kidoq, which allows the prediction of Ki value of a new ligand molecule against DHDPS.
鉴定新的药物靶点及其抑制剂是药物设计和开发领域的主要挑战。二氨基庚二酸(DAP)途径是一种存在于细菌中的独特赖氨酸生物合成途径,而在哺乳动物中不存在。该途径对细菌至关重要,因为它在细胞壁生物合成中起着关键作用。该途径的一个重要酶是二氢二吡啶羧酸合酶(DHDPS),被认为对细菌的生存至关重要。鉴于其重要性,开发和预测针对 DHDPS 的有效抑制剂可能有助于设计针对细菌的有效药物。
本文描述了一种针对 DHDPS 的新型/有效抑制剂预测方法。在这里,使用实验验证的 23 种酶抑制剂(Ki 值在 0.005-22mM 范围内)对定量构效关系(QSAR)模型进行了训练和测试。这些抑制剂使用 AutoDock 软件在 DHDPS(1YXD)的活性部位进行对接,得到 11 个基于能量的描述符。对于 QSAR 建模,使用最佳的四个基于能量的描述符生成了多元线性回归(MLR)模型,其相关值 R/q2 为 0.82/0.67,MAE 为 2.43。此外,使用 F-逐步消除方法选择三个关键描述符,开发了基于支持向量机(SVM)的模型,从而提高了性能,达到了 R/q2 值为 0.93/0.80 和 MAE 为 1.89。为了验证 QSAR 模型的性能,采用了外部交叉验证程序,该程序在 0.78-0.83/0.93-0.95 的范围内实现了高的训练/测试相关值(q2/r2)。
我们的结果表明,使用 QSAR 建模研究 DHDPS 的配体-受体结合相互作用似乎是一种有前途的预测抗菌剂的方法。为了帮助实验人员开发新型/有效抑制剂,我们开发了一个名为“KiDoQ”的网络服务器 http://crdd.osdd.net/raghava/kidoq,该服务器可以预测新的配体分子对 DHDPS 的 Ki 值。