Center for Comparative Genomics and Bioinformatics, Pennsylvania State University, USA.
BMC Bioinformatics. 2010 Mar 15;11:130. doi: 10.1186/1471-2105-11-130.
The most common application for the next-generation sequencing technologies is resequencing, where short reads from the genome of an individual are aligned to a reference genome sequence for the same species. These mappings can then be used to identify genetic differences among individuals in a population, and perhaps ultimately to explain phenotypic variation. Many algorithms capable of aligning short reads to the reference, and determining differences between them have been reported. Much less has been reported on how to use these technologies to determine genetic differences among individuals of a species for which a reference sequence is not available, which drastically limits the number of species that can easily benefit from these new technologies.
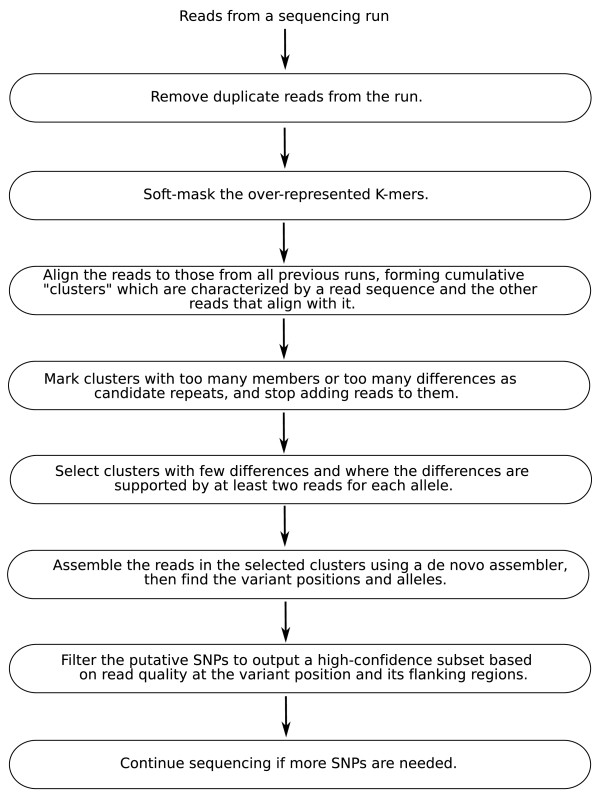
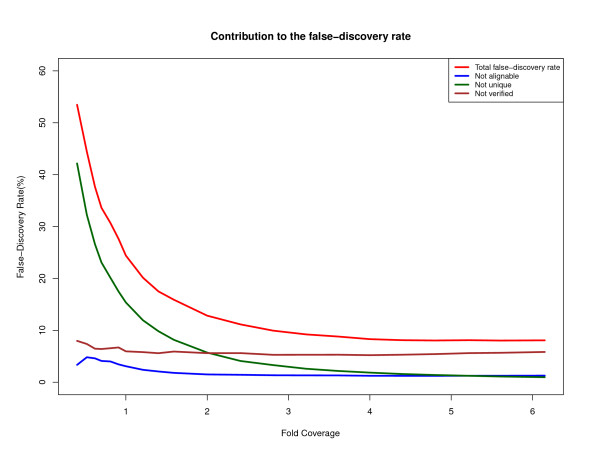
We describe a computational pipeline, called DIAL (De novo Identification of Alleles), for identifying single-base substitutions between two closely related genomes without the help of a reference genome. The method works even when the depth of coverage is insufficient for de novo assembly, and it can be extended to determine small insertions/deletions. We evaluate the software's effectiveness using published Roche/454 sequence data from the genome of Dr. James Watson (to detect heterozygous positions) and recent Illumina data from orangutan, in each case comparing our results to those from computational analysis that uses a reference genome assembly. We also illustrate the use of DIAL to identify nucleotide differences among transcriptome sequences.
DIAL can be used for identification of nucleotide differences in species for which no reference sequence is available. Our main motivation is to use this tool to survey the genetic diversity of endangered species as the identified sequence differences can be used to design genotyping arrays to assist in the species' management. The DIAL source code is freely available at http://www.bx.psu.edu/miller_lab/.
下一代测序技术最常见的应用是重测序,即将个体基因组的短读段与同一物种的参考基因组序列进行比对。这些比对可以用来识别群体中个体之间的遗传差异,并最终解释表型变异。已经有许多能够将短读段与参考序列进行比对并确定它们之间差异的算法被报道。但关于如何利用这些技术来确定没有参考序列的物种中个体之间的遗传差异的报道却很少,这极大地限制了可以从这些新技术中受益的物种数量。
我们描述了一种名为 DIAL(从头鉴定等位基因)的计算流程,用于在没有参考基因组的情况下识别两个密切相关的基因组之间的单碱基替换。该方法甚至在覆盖深度不足以进行从头组装的情况下也能工作,并且可以扩展用于确定小的插入/缺失。我们使用已发表的 Roche/454 序列数据(来自 Dr. James Watson 的基因组,用于检测杂合位置)和最近的猩猩 Illumina 数据来评估软件的有效性,在每种情况下,我们将结果与使用参考基因组组装的计算分析进行比较。我们还展示了 DIAL 用于鉴定转录组序列中核苷酸差异的用法。
DIAL 可用于鉴定没有参考序列的物种中的核苷酸差异。我们的主要动机是使用此工具来调查濒危物种的遗传多样性,因为所鉴定的序列差异可用于设计基因分型阵列以协助物种管理。DIAL 的源代码可在 http://www.bx.psu.edu/miller_lab/ 上免费获得。