Kerstens Hindrik H D, Crooijmans Richard P M A, Veenendaal Albertine, Dibbits Bert W, Chin-A-Woeng Thomas F C, den Dunnen Johan T, Groenen Martien A M
Animal Breeding and Genomics Center, Wageningen University, Marijkeweg 40, Wageningen, 6709 PG, the Netherlands.
BMC Genomics. 2009 Oct 16;10:479. doi: 10.1186/1471-2164-10-479.
The development of second generation sequencing methods has enabled large scale DNA variation studies at moderate cost. For the high throughput discovery of single nucleotide polymorphisms (SNPs) in species lacking a sequenced reference genome, we set-up an analysis pipeline based on a short read de novo sequence assembler and a program designed to identify variation within short reads. To illustrate the potential of this technique, we present the results obtained with a randomly sheared, enzymatically generated, 2-3 kbp genome fraction of six pooled Meleagris gallopavo (turkey) individuals.
A total of 100 million 36 bp reads were generated, representing approximately 5-6% (approximately 62 Mbp) of the turkey genome, with an estimated sequence depth of 58. Reads consisting of bases called with less than 1% error probability were selected and assembled into contigs. Subsequently, high throughput discovery of nucleotide variation was performed using sequences with more than 90% reliability by using the assembled contigs that were 50 bp or longer as the reference sequence. We identified more than 7,500 SNPs with a high probability of representing true nucleotide variation in turkeys. Increasing the reference genome by adding publicly available turkey BAC-end sequences increased the number of SNPs to over 11,000. A comparison with the sequenced chicken genome indicated that the assembled turkey contigs were distributed uniformly across the turkey genome. Genotyping of a representative sample of 340 SNPs resulted in a SNP conversion rate of 95%. The correlation of the minor allele count (MAC) and observed minor allele frequency (MAF) for the validated SNPs was 0.69.
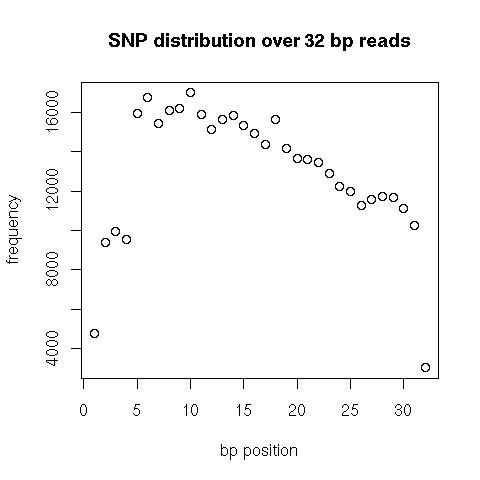
We provide an efficient and cost-effective approach for the identification of thousands of high quality SNPs in species currently lacking a sequenced genome and applied this to turkey. The methodology addresses a random fraction of the genome, resulting in an even distribution of SNPs across the targeted genome.
第二代测序方法的发展使得大规模DNA变异研究能够以适中的成本进行。为了在缺乏已测序参考基因组的物种中高通量发现单核苷酸多态性(SNP),我们基于短读长从头序列组装器和一个用于识别短读长内变异的程序建立了一个分析流程。为了说明该技术的潜力,我们展示了对六个混合的火鸡个体随机剪切、酶促产生的2 - 3kbp基因组片段进行分析所获得的结果。
共产生了1亿条36bp的读段,约占火鸡基因组的5 - 6%(约62Mbp),估计序列深度为58。选择错误概率小于1%的碱基组成的读段并组装成重叠群。随后,使用可靠性超过90%的序列,以50bp或更长的组装重叠群作为参考序列,进行核苷酸变异的高通量发现。我们鉴定出超过7500个SNP,它们极有可能代表火鸡中的真实核苷酸变异。通过添加公开可用的火鸡BAC末端序列来增加参考基因组,使SNP数量增加到超过11000个。与已测序的鸡基因组进行比较表明,组装的火鸡重叠群在火鸡基因组中均匀分布。对340个SNP的代表性样本进行基因分型,SNP转化率为95%。验证后的SNP的次要等位基因计数(MAC)与观察到的次要等位基因频率(MAF)的相关性为0.69。
我们提供了一种高效且经济有效的方法,用于在目前缺乏已测序基因组的物种中鉴定数千个高质量SNP,并将其应用于火鸡。该方法针对基因组的随机部分,导致SNP在目标基因组中均匀分布。