Department of Epidemiology and Public Health, National University of Singapore, 16 Medical Drive, Singapore.
BMC Bioinformatics. 2010 Mar 22;11:147. doi: 10.1186/1471-2105-11-147.
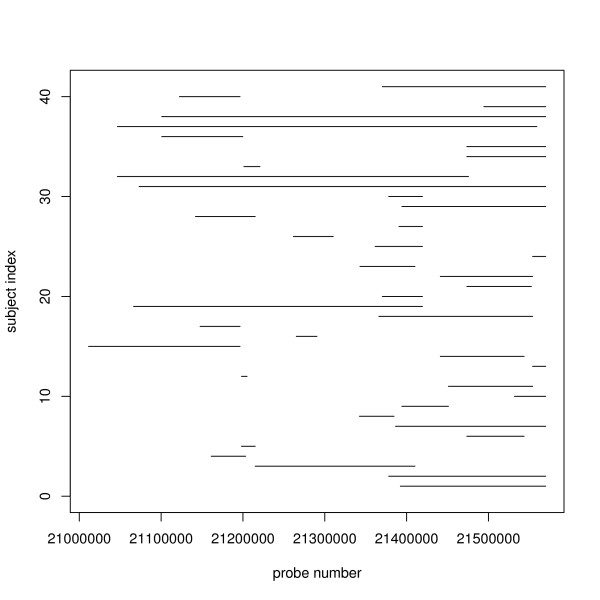
Algorithms and software for CNV detection have been developed, but they detect the CNV regions sample-by-sample with individual-specific breakpoints, while common CNV regions are likely to occur at the same genomic locations across different individuals in a homogenous population. Current algorithms to detect common CNV regions do not account for the varying reliability of the individual CNVs, typically reported as confidence scores by SNP-based CNV detection algorithms. General methodologies for identifying these recurrent regions, especially those directed at SNP arrays, are still needed.
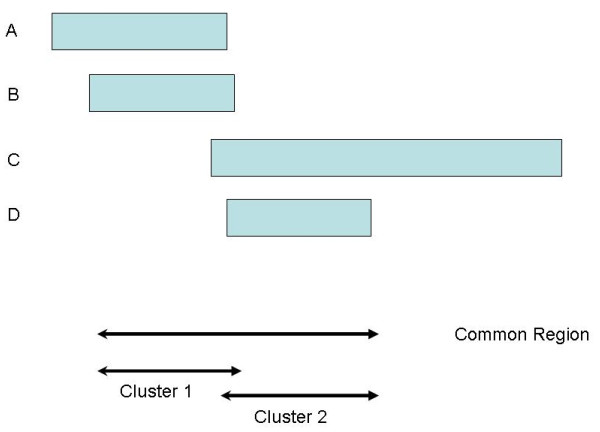
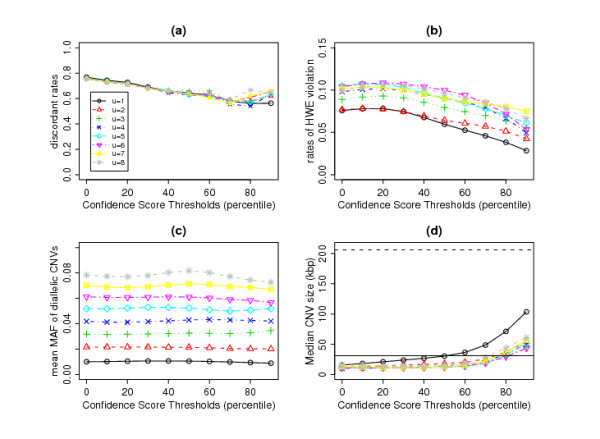
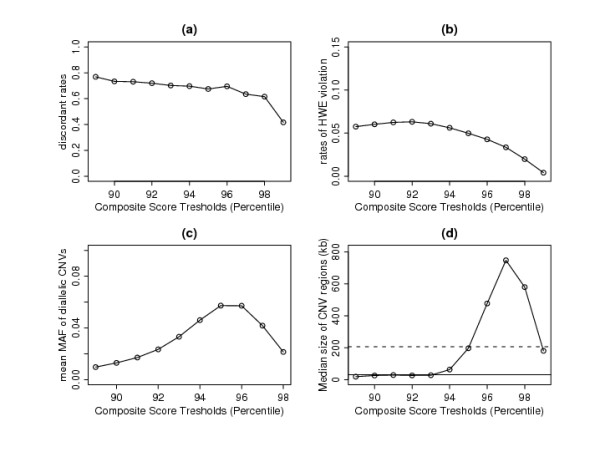
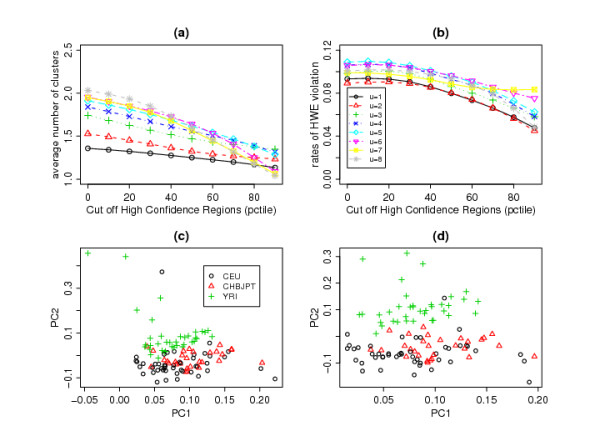
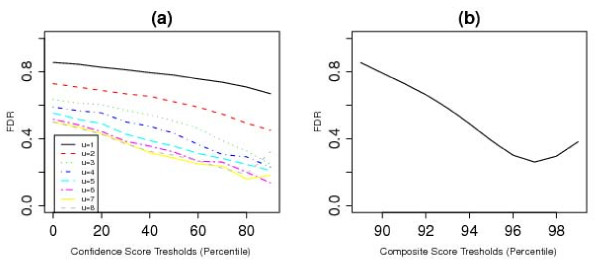
In this paper, we describe two new approaches for identifying common CNV regions based on (i) the frequency of occurrence of reliable CNVs, where reliability is determined by high confidence scores, and (ii) a weighted frequency of occurrence of CNVs, where the weights are determined by the confidence scores. In addition, motivated by the fact that we often observe partially overlapping CNV regions as a mixture of two or more distinct subregions, regions identified using the two approaches can be fine-tuned to smaller sub-regions using a clustering algorithm. We compared the performance of the methods with sequencing-based results in terms of discordance rates, rates of departure from Hardy-Weinberg equilibrium (HWE) and average frequency and size of the identified regions. The discordance rates as well as the rates of departure from HWE decrease when we select CNVs with higher confidence scores. We also performed comparisons with two previously published methods, STAC and GISTIC, and showed that the methods we consider are better at identifying low-frequency but high-confidence CNV regions.
The proposed methods for identifying common CNV regions in multiple individuals perform well compared to existing methods. The identified common regions can be used for downstream analyses such as group comparisons in association studies.
已经开发出用于 CNV 检测的算法和软件,但它们逐个样本地检测 CNV 区域,具有个体特异性断点,而常见的 CNV 区域可能在同质人群的不同个体中出现在相同的基因组位置。当前用于检测常见 CNV 区域的算法没有考虑到个体 CNV 的变化可靠性,通常 SNP 基 CNV 检测算法报告为置信分数。仍需要确定这些重复区域的一般方法,特别是针对 SNP 阵列的方法。
在本文中,我们描述了两种基于(i)可靠 CNV 发生频率的识别常见 CNV 区域的新方法,其中可靠性由高置信分数确定,以及(ii)CNV 发生频率的加权频率确定,其中权重由置信分数确定。此外,鉴于我们经常观察到部分重叠的 CNV 区域作为两个或更多不同子区域的混合物,因此可以使用聚类算法将使用两种方法识别的区域调整为较小的子区域。我们比较了这些方法与基于测序的结果在不一致率、偏离 Hardy-Weinberg 平衡(HWE)的比率以及识别区域的平均频率和大小方面的性能。当我们选择具有更高置信分数的 CNV 时,不一致率以及偏离 HWE 的比率会降低。我们还与两种先前发表的方法 STAC 和 GISTIC 进行了比较,并表明我们考虑的方法在识别低频但高置信度的 CNV 区域方面表现更好。
与现有方法相比,用于在多个个体中识别常见 CNV 区域的提出的方法表现良好。鉴定的常见区域可用于下游分析,例如关联研究中的组比较。