Delft Bioinformatics Lab, Delft University of Technology, Mekelweg 4, Delft 2628 CD, The Netherlands.
BMC Bioinformatics. 2010 Mar 26;11:156. doi: 10.1186/1471-2105-11-156.
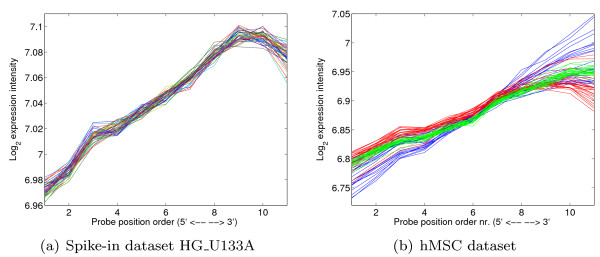
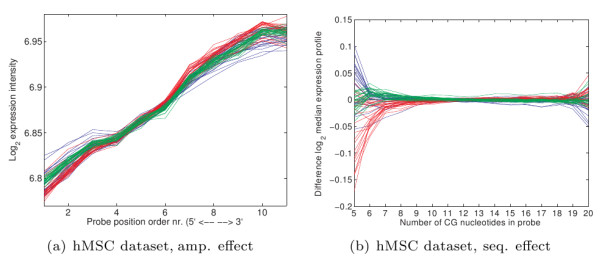
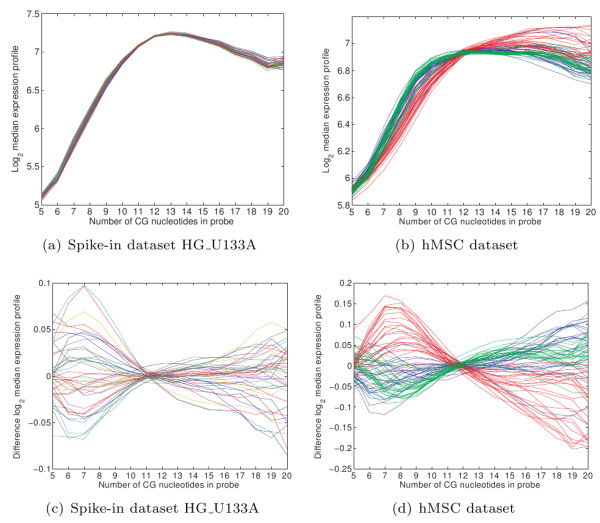
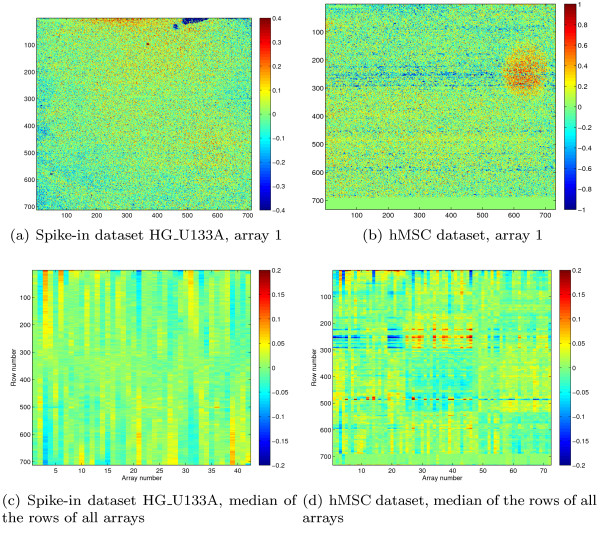
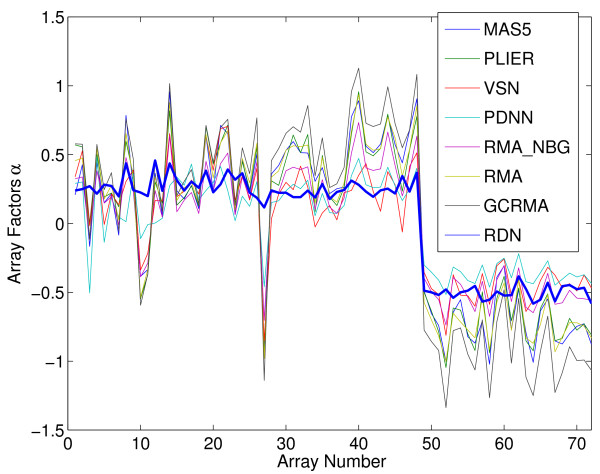
Oligonucleotide arrays have become one of the most widely used high-throughput tools in biology. Due to their sensitivity to experimental conditions, normalization is a crucial step when comparing measurements from these arrays. Normalization is, however, far from a solved problem. Frequently, we encounter datasets with significant technical effects that currently available methods are not able to correct.
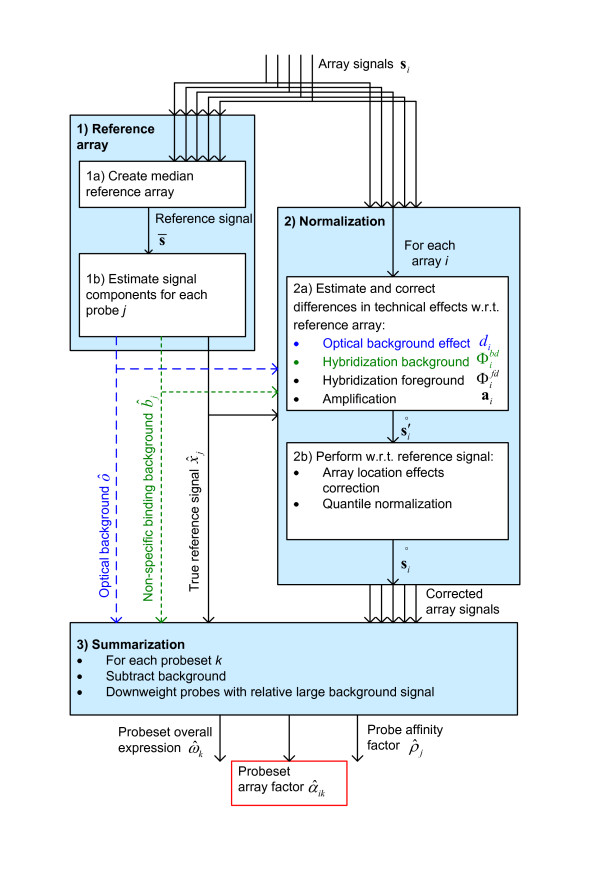
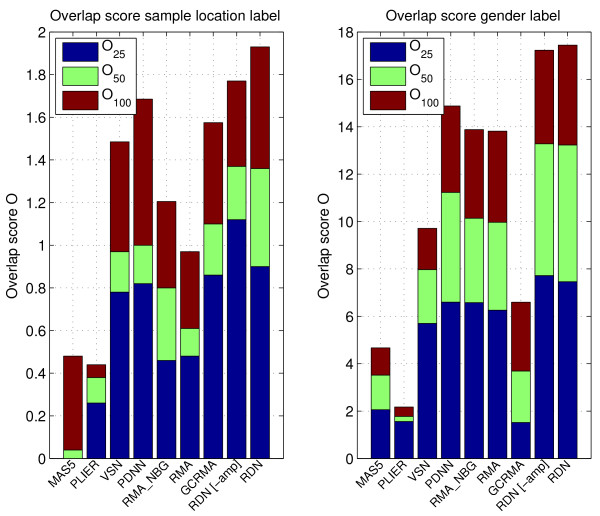
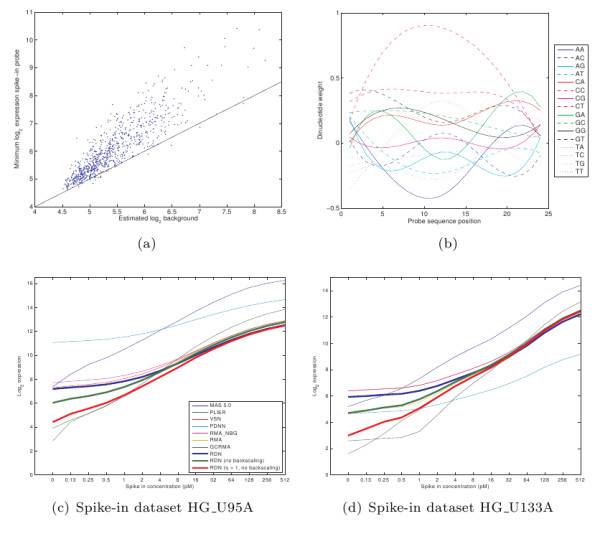
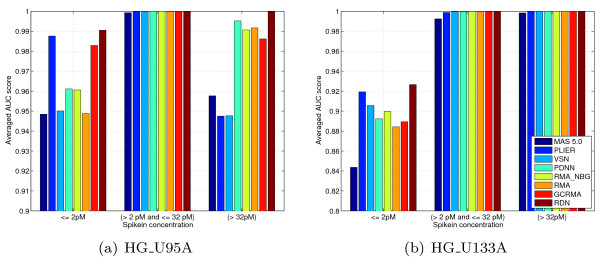
We show that by a careful decomposition of probe specific amplification, hybridization and array location effects, a normalization can be performed that allows for a much improved analysis of these data. Identification of the technical sources of variation between arrays has allowed us to build statistical models that are used to estimate how the signal of individual probes is affected, based on their properties. This enables a model-based normalization that is probe-specific, in contrast with the signal intensity distribution normalization performed by many current methods. Next to this, we propose a novel way of handling background correction, enabling the use of background information to weight probes during summarization. Testing of the proposed method shows a much improved detection of differentially expressed genes over earlier proposed methods, even when tested on (experimentally tightly controlled and replicated) spike-in datasets.
When a limited number of arrays are available, or when arrays are run in different batches, technical effects have a large influence on the measured expression of genes. We show that a detailed modelling and correction of these technical effects allows for an improved analysis in these situations.
寡核苷酸微阵列已成为生物学中应用最广泛的高通量工具之一。由于它们对实验条件敏感,因此在比较这些微阵列的测量值时,归一化是一个关键步骤。然而,归一化远未得到解决。我们经常遇到具有显著技术效应的数据集,而目前可用的方法无法纠正这些效应。
我们通过仔细分解探针特异性扩增、杂交和微阵列位置效应,可以进行归一化,从而大大改进对这些数据的分析。鉴定微阵列之间技术变异的来源使我们能够构建统计模型,根据探针的特性来估计单个探针的信号如何受到影响。这使得可以进行基于模型的归一化,与许多当前方法执行的信号强度分布归一化形成对比。除此之外,我们还提出了一种新的背景校正处理方法,在汇总时可以使用背景信息来加权探针。对所提出方法的测试表明,即使在(经过严格实验控制和复制的) Spike-in 数据集上进行测试,与早期提出的方法相比,该方法在检测差异表达基因方面有了很大的改进。
当可用的微阵列数量有限,或者微阵列在不同批次中运行时,技术效应会对基因的测量表达产生很大影响。我们表明,详细建模和纠正这些技术效应可以在这些情况下改善分析。