Institut Curie, 26 rue d'Ulm, Paris, France.
Nucleic Acids Res. 2010 Jun;38(11):e126. doi: 10.1093/nar/gkq217. Epub 2010 Apr 7.
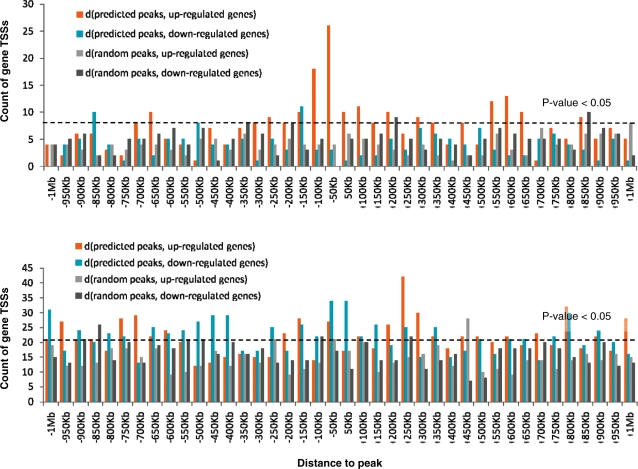
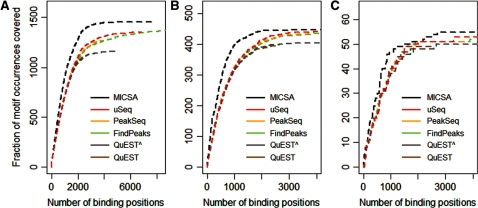
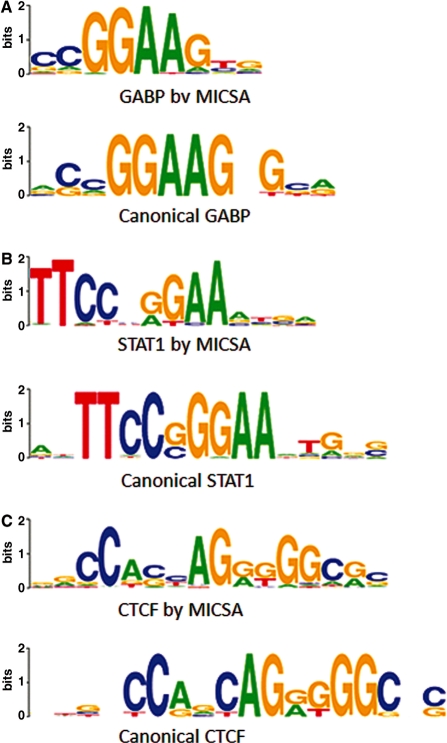
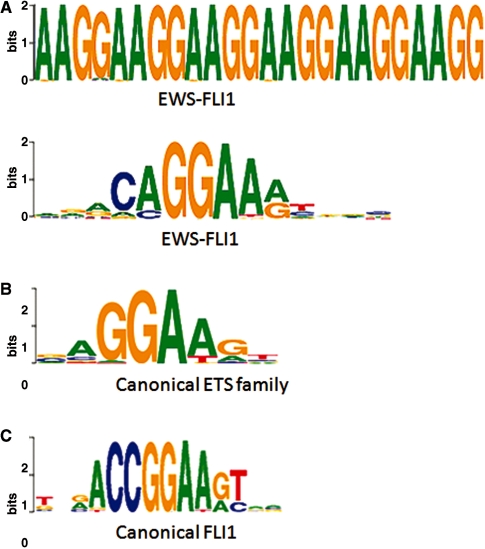
Dramatic progress in the development of next-generation sequencing technologies has enabled accurate genome-wide characterization of the binding sites of DNA-associated proteins. This technique, baptized as ChIP-Seq, uses a combination of chromatin immunoprecipitation and massively parallel DNA sequencing. Other published tools that predict binding sites from ChIP-Seq data use only positional information of mapped reads. In contrast, our algorithm MICSA (Motif Identification for ChIP-Seq Analysis) combines this source of positional information with information on motif occurrences to better predict binding sites of transcription factors (TFs). We proved the greater accuracy of MICSA with respect to several other tools by running them on datasets for the TFs NRSF, GABP, STAT1 and CTCF. We also applied MICSA on a dataset for the oncogenic TF EWS-FLI1. We discovered >2000 binding sites and two functionally different binding motifs. We observed that EWS-FLI1 can activate gene transcription when (i) its binding site is located in close proximity to the gene transcription start site (up to approximately 150 kb), and (ii) it contains a microsatellite sequence. Furthermore, we observed that sites without microsatellites can also induce regulation of gene expression--positively as often as negatively--and at much larger distances (up to approximately 1 Mb).
下一代测序技术的飞速发展使得对 DNA 相关蛋白的结合位点进行全基因组精确描绘成为可能。这种技术被称为 ChIP-Seq,它结合了染色质免疫沉淀和大规模平行 DNA 测序。其他从 ChIP-Seq 数据预测结合位点的已发表工具仅使用映射读取的位置信息。相比之下,我们的算法 MICSA(ChIP-Seq 分析中的基序识别)将这种位置信息与基序出现的信息相结合,以更好地预测转录因子 (TF) 的结合位点。我们通过在 NRSF、GABP、STAT1 和 CTCF 的 TF 数据集上运行这些工具,证明了 MICSA 相对于其他几个工具具有更高的准确性。我们还将 MICSA 应用于致癌 TF EWS-FLI1 的数据集。我们发现了 >2000 个结合位点和两个功能不同的结合基序。我们观察到,当 EWS-FLI1 的结合位点 (i) 位于基因转录起始位点附近(最多约 150kb),和 (ii) 包含微卫星序列时,它可以激活基因转录。此外,我们还观察到没有微卫星的位点也可以诱导基因表达的调控——积极的和消极的——并且在更大的距离(最多约 1Mb)。