Department of Intelligence Science and Technology, Graduate School of Informatics, Kyoto University, Yoshida-Honmachi, Sakyo-ku, Kyoto-shi, Kyoto 606-8501, Japan.
BMC Bioinformatics. 2010 Apr 30;11:224. doi: 10.1186/1471-2105-11-224.
Whole-genome sequence alignment is an essential process for extracting valuable information about the functions, evolution, and peculiarities of genomes under investigation. As available genomic sequence data accumulate rapidly, there is great demand for tools that can compare whole-genome sequences within practical amounts of time and space. However, most existing genomic alignment tools can treat sequences that are only a few Mb long at once, and no state-of-the-art alignment program can align large sequences such as mammalian genomes directly on a conventional standalone computer.
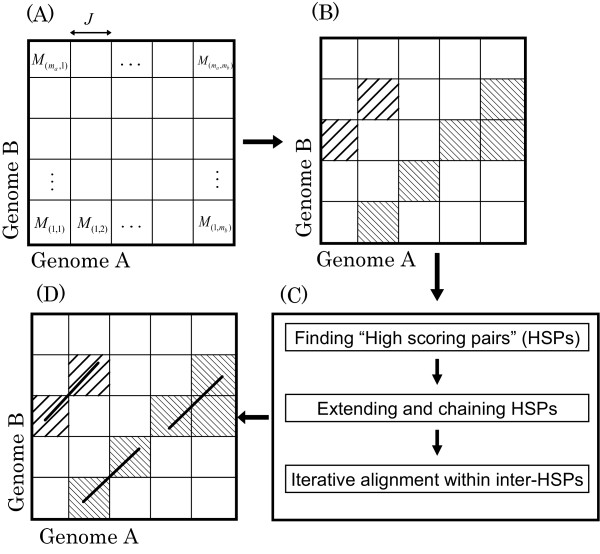
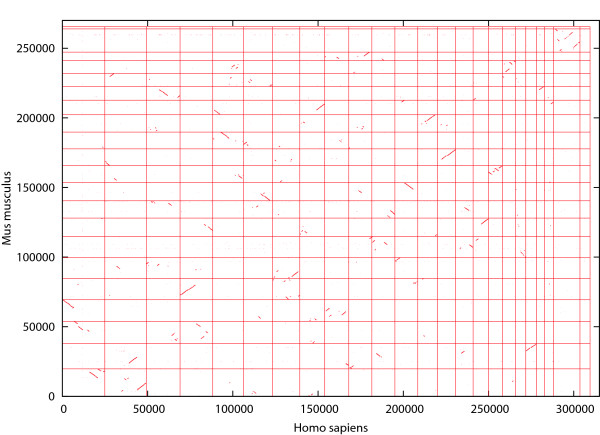
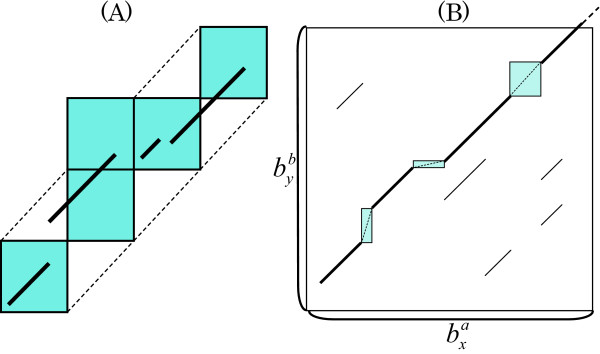
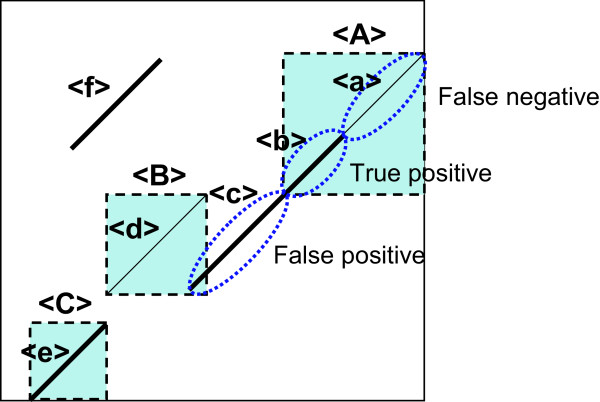
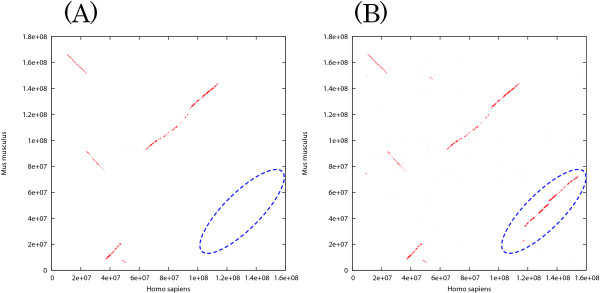
We previously proposed the CGAT (Coarse-Grained AlignmenT) algorithm, which performs an alignment job in two steps: first at the block level and then at the nucleotide level. The former is "coarse-grained" alignment that can explore genomic rearrangements and reduce the sizes of the regions to be analyzed in the next step. The latter is detailed alignment within limited regions. In this paper, we present an update of the algorithm and the open-source program, Cgaln, that implements the algorithm. We compared the performance of Cgaln with those of other programs on whole genomic sequences of several bacteria and of some mammalian chromosome pairs. The results showed that Cgaln is several times faster and more memory-efficient than the best existing programs, while its sensitivity and accuracy are comparable to those of the best programs. Cgaln takes less than 13 hours to finish an alignment between the whole genomes of human and mouse in a single run on a conventional desktop computer with a single CPU and 2 GB memory.
Cgaln is not only fast and memory efficient but also effective in coping with genomic rearrangements. Our results show that Cgaln is very effective for comparison of large genomes, especially of intact chromosomal sequences. We believe that Cgaln provides novel viewpoint for reducing computational complexity and will contribute to various fields of genome science.
全基因组序列比对是从研究中的基因组功能、进化和特征中提取有价值信息的必要过程。随着可用基因组序列数据的快速积累,人们对能够在实际的时间和空间内比较全基因组序列的工具产生了巨大的需求。然而,大多数现有的基因组比对工具一次只能处理几 Mb 长的序列,并且没有最先进的对齐程序可以直接在传统的独立计算机上对齐大型序列,如哺乳动物基因组。
我们之前提出了 CGAT(粗粒度对齐)算法,该算法分两步执行对齐工作:首先在块级别,然后在核苷酸级别。前者是“粗粒度”对齐,可以探索基因组重排并减少下一步要分析的区域的大小。后者是在有限区域内的详细对齐。在本文中,我们介绍了该算法的更新和开源程序 Cgaln,该程序实现了该算法。我们将 Cgaln 的性能与其他程序在几种细菌的全基因组序列和一些哺乳动物染色体对上的性能进行了比较。结果表明,Cgaln 的速度比现有最好的程序快几倍,内存效率更高,而其敏感性和准确性与最好的程序相当。Cgaln 在单个 CPU 和 2GB 内存的传统台式计算机上单个运行时,完成人类和小鼠全基因组之间的对齐不到 13 小时。
Cgaln 不仅速度快、内存效率高,而且能有效地处理基因组重排。我们的结果表明,Cgaln 非常有效地用于比较大型基因组,特别是完整的染色体序列。我们相信 Cgaln 为降低计算复杂性提供了新的视角,并将为基因组科学的各个领域做出贡献。