Ontario Institute for Cancer Research, MaRS Centre, South Tower, 101 College Street, Suite 800, Toronto, ON M5G 0A3, Canada.
Genome Biol. 2010;11(5):R53. doi: 10.1186/gb-2010-11-5-r53. Epub 2010 May 19.
One challenge facing biologists is to tease out useful information from massive data sets for further analysis. A pathway-based analysis may shed light by projecting candidate genes onto protein functional relationship networks. We are building such a pathway-based analysis system.
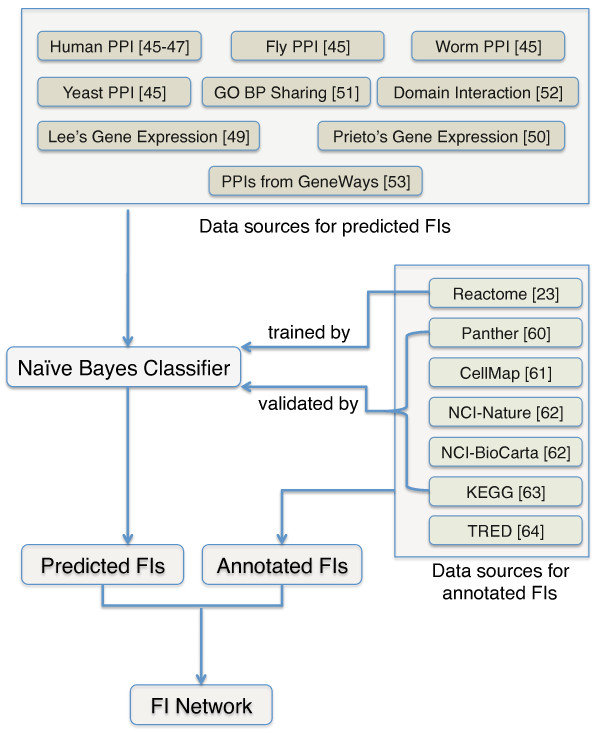
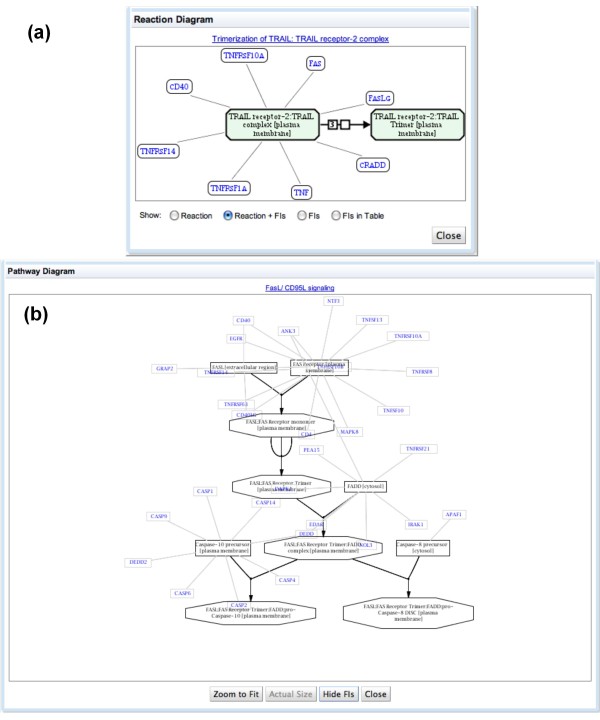
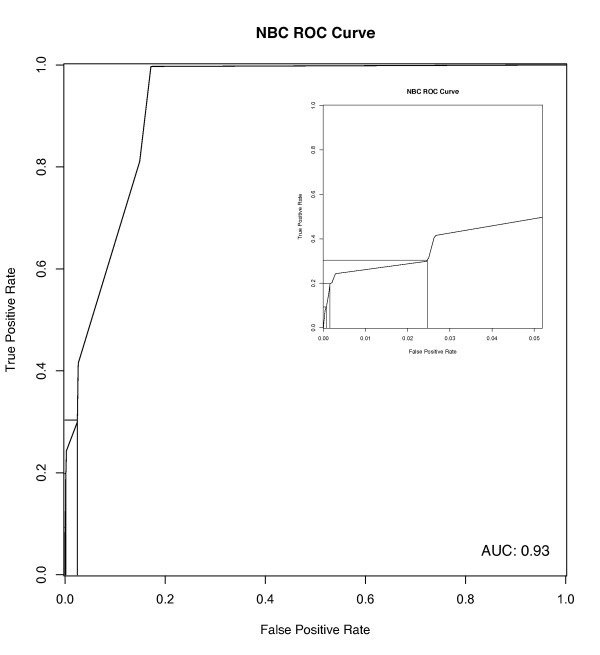
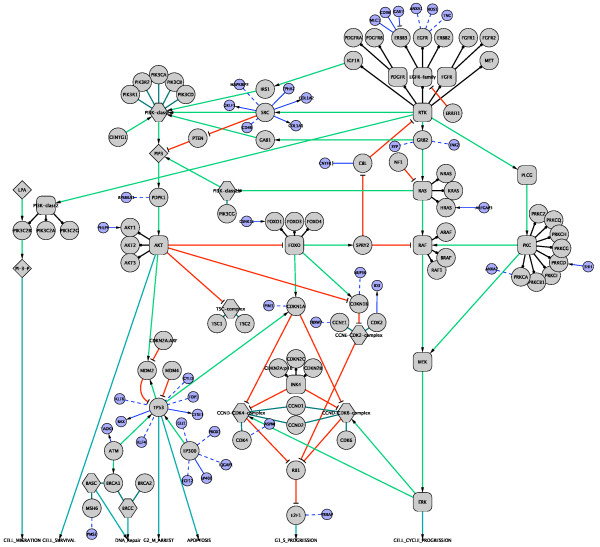
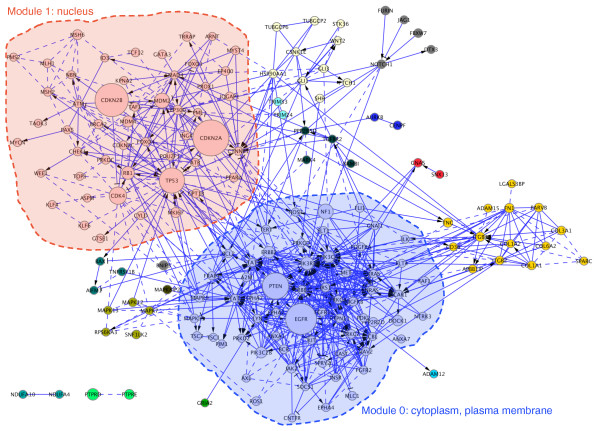
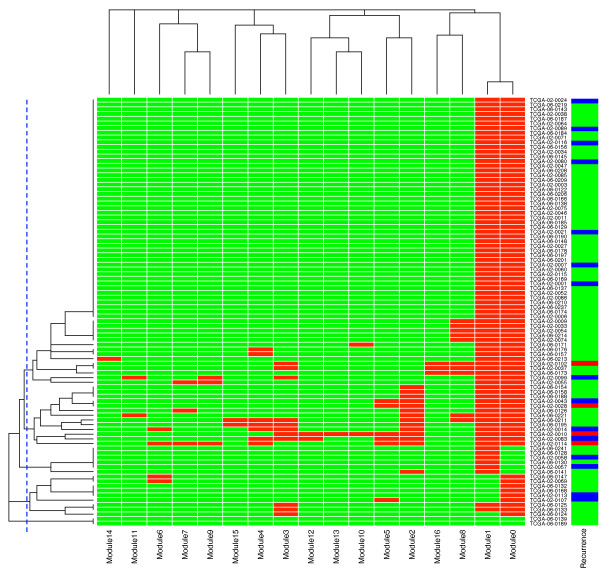
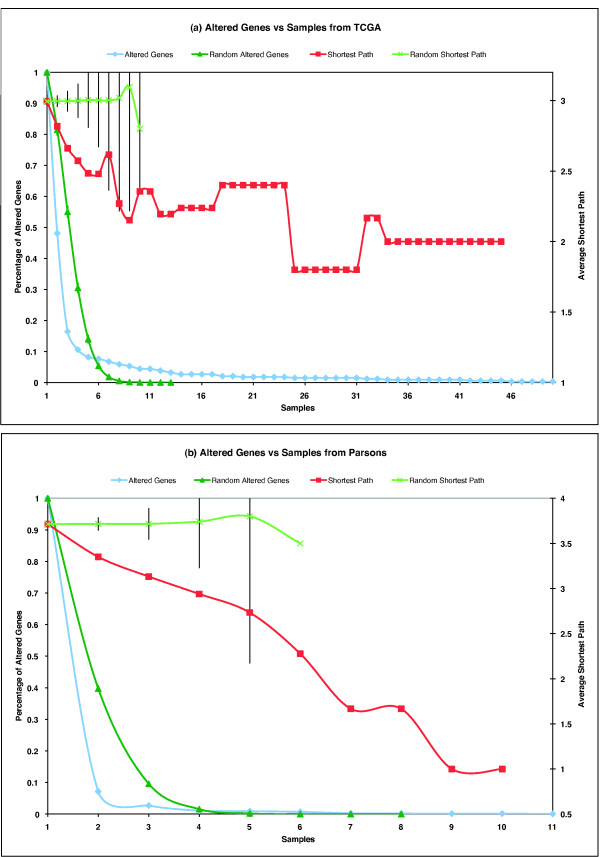
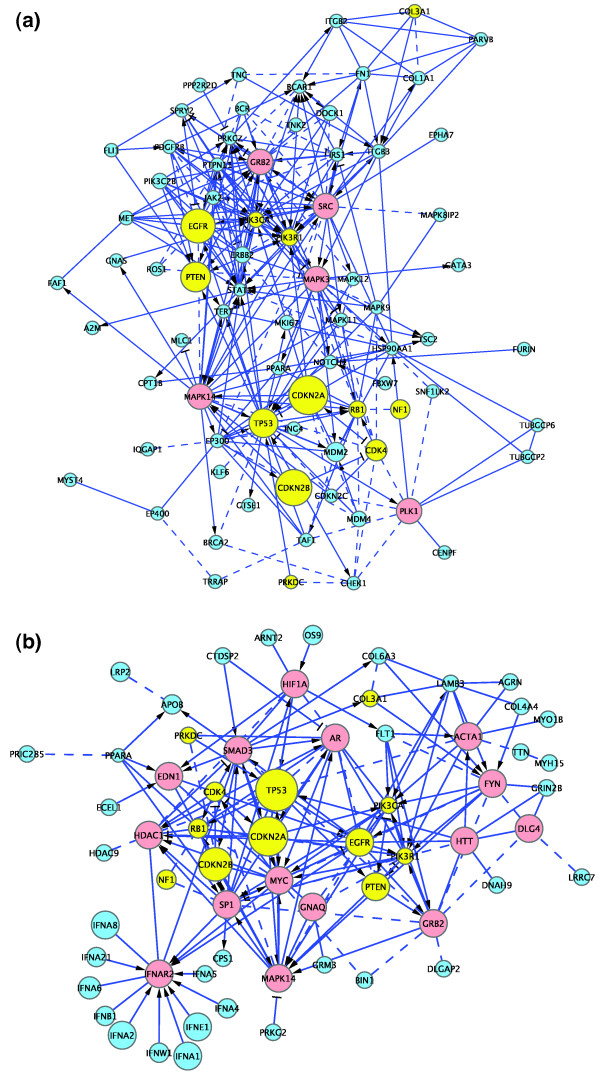
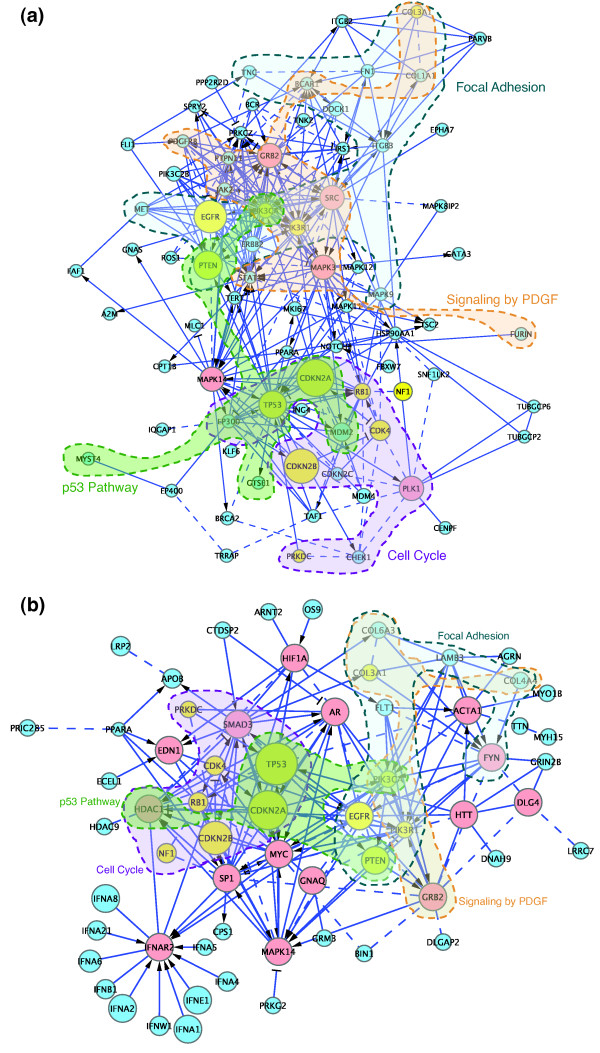
We have constructed a protein functional interaction network by extending curated pathways with non-curated sources of information, including protein-protein interactions, gene coexpression, protein domain interaction, Gene Ontology (GO) annotations and text-mined protein interactions, which cover close to 50% of the human proteome. By applying this network to two glioblastoma multiforme (GBM) data sets and projecting cancer candidate genes onto the network, we found that the majority of GBM candidate genes form a cluster and are closer than expected by chance, and the majority of GBM samples have sequence-altered genes in two network modules, one mainly comprising genes whose products are localized in the cytoplasm and plasma membrane, and another comprising gene products in the nucleus. Both modules are highly enriched in known oncogenes, tumor suppressors and genes involved in signal transduction. Similar network patterns were also found in breast, colorectal and pancreatic cancers.
We have built a highly reliable functional interaction network upon expert-curated pathways and applied this network to the analysis of two genome-wide GBM and several other cancer data sets. The network patterns revealed from our results suggest common mechanisms in the cancer biology. Our system should provide a foundation for a network or pathway-based analysis platform for cancer and other diseases.
生物学家面临的一个挑战是从大量数据集提取有用信息以进行进一步分析。基于途径的分析可以通过将候选基因投射到蛋白质功能关系网络上来提供启示。我们正在构建这样一个基于途径的分析系统。
我们通过扩展经过精心整理的途径,结合非精心整理的信息来源,构建了一个蛋白质功能相互作用网络,包括蛋白质-蛋白质相互作用、基因共表达、蛋白质结构域相互作用、基因本体 (GO) 注释和文本挖掘的蛋白质相互作用,这些信息涵盖了近 50%的人类蛋白质组。通过将该网络应用于两个胶质母细胞瘤多形性 (GBM) 数据集,并将癌症候选基因投射到网络上,我们发现大多数 GBM 候选基因形成一个簇,比随机预期的更接近,并且大多数 GBM 样本在两个网络模块中都有序列改变的基因,一个主要包含其产物定位于细胞质和质膜的基因,另一个包含核内基因产物的基因。这两个模块都高度富含已知的癌基因、肿瘤抑制基因和参与信号转导的基因。在乳腺癌、结直肠癌和胰腺癌中也发现了类似的网络模式。
我们在专家整理的途径基础上构建了一个高度可靠的功能相互作用网络,并将该网络应用于两个全基因组 GBM 和其他几个癌症数据集的分析。我们从结果中揭示的网络模式表明癌症生物学中存在共同的机制。我们的系统应为癌症和其他疾病的网络或途径分析平台提供基础。