Department of Medicine, Washington University, St Louis, MO 63110, USA.
Circ Res. 2010 Jul 23;107(2):271-82. doi: 10.1161/CIRCRESAHA.110.219899. Epub 2010 May 20.
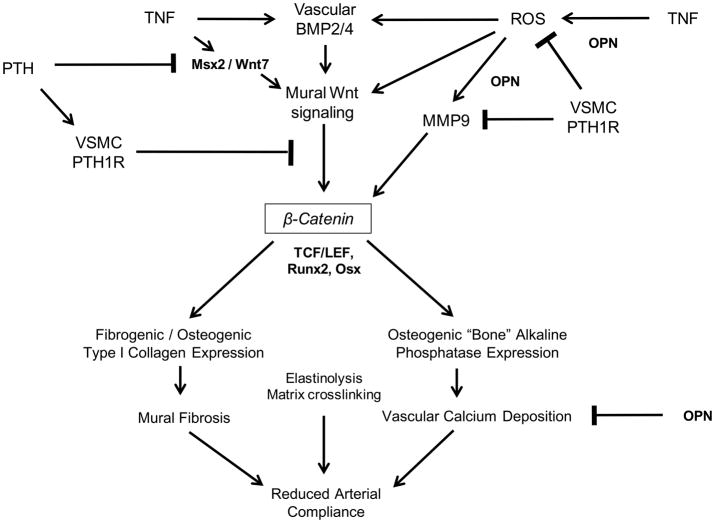
Vascular fibrosis and calcification contribute to diabetic arteriosclerosis, impairing Windkessel physiology necessary for distal tissue perfusion. Wnt family members, upregulated in arteries by the low-grade inflammation of "diabesity," stimulate type I collagen expression and osteogenic mineralization of mesenchymal progenitors via beta-catenin. Conversely, parathyroid hormone (PTH) inhibits aortic calcification in low-density lipoprotein receptor (LDLR)-deficient mice fed high fat diabetogenic diets (HFD).
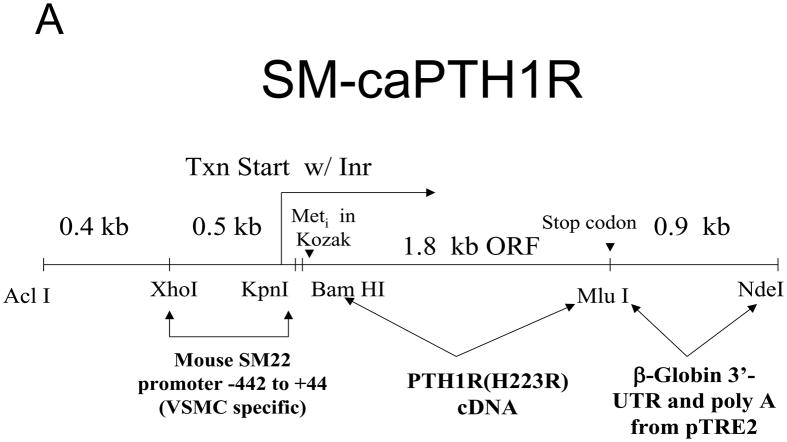
We sought to determine the impact of vascular PTH receptor (PTH1R) activity on arteriosclerotic Wnt/beta-catenin signaling in vitro and in vivo. We generated SM-caPTH1R transgenic mice, a model in which the constitutively active PTH1R variant H223R (caPTH1R) is expressed only in the vasculature.
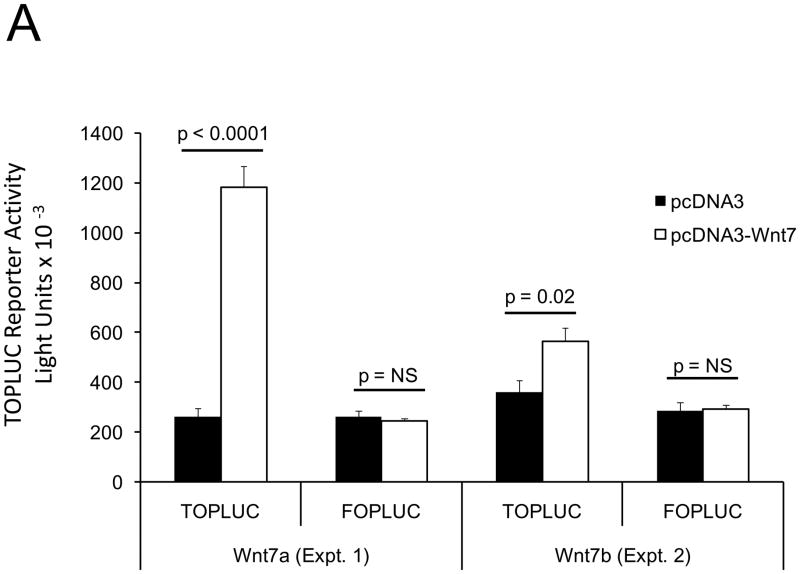
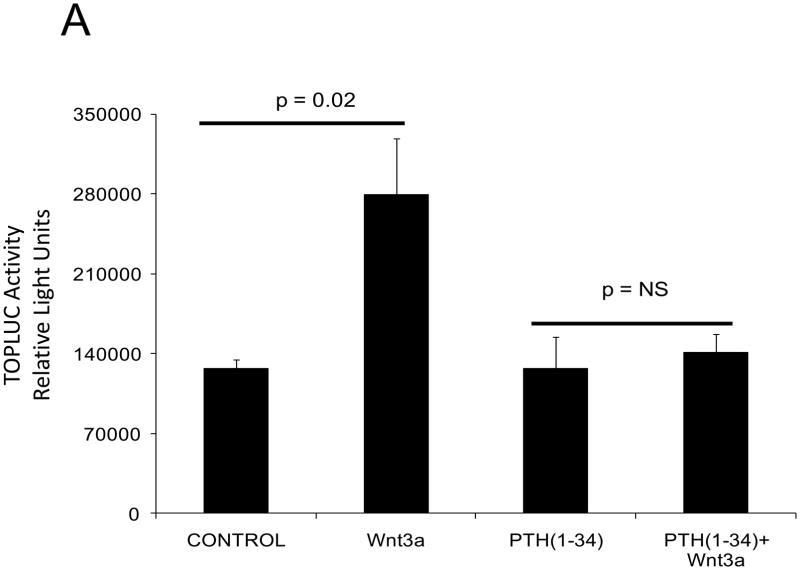
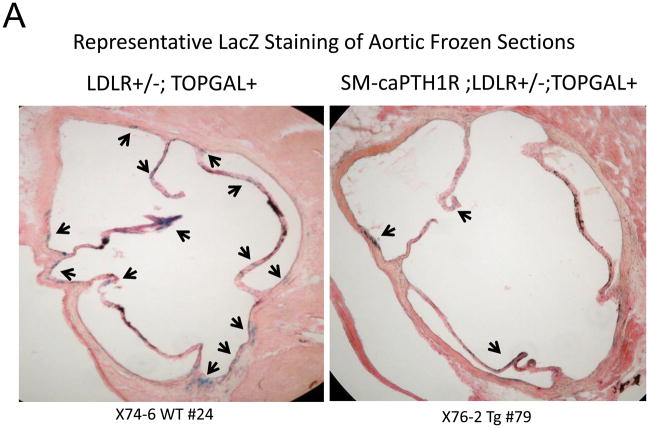
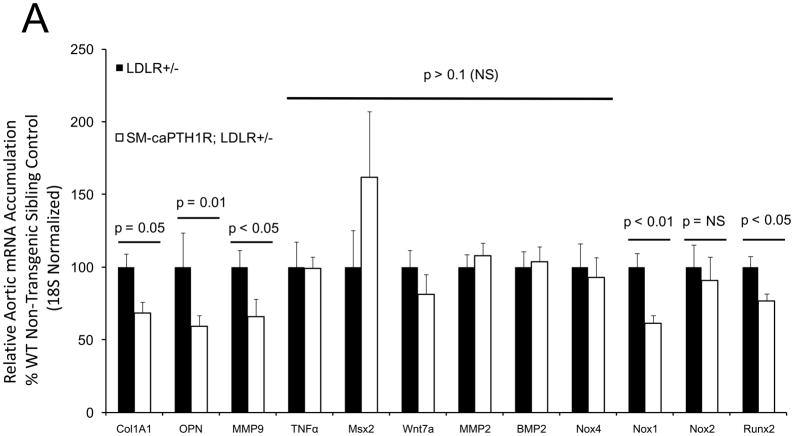
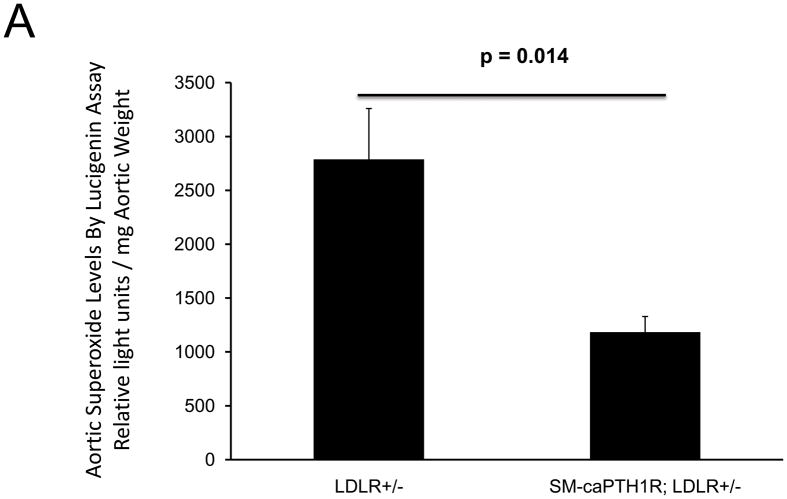
The caPTH1R inhibited Wnt/beta-catenin signaling, collagen production, and vascular smooth muscle cell proliferation and calcification in vitro. Transgenic SM-caPTH1R;LDLR(+/-) mice fed HFD develop diabesity, with no improvements in fasting serum glucose, cholesterol, weight, body composition, or bone mass versus LDLR(+/-) siblings. SM-caPTH1R downregulated aortic Col1A1, Runx2, and Nox1 expression without altering TNF, Msx2, Wnt7a/b, or Nox4. The SM-caPTH1R transgene decreased aortic beta-catenin protein accumulation and signaling in diabetic LDLR(+/-) mice. Levels of aortic superoxide (a precursor of peroxide that activates pro-matrix metalloproteinase 9 and osteogenic signaling in vascular smooth muscle cells) were suppressed by the SM-caPTH1R transgene. Aortic calcification, collagen accumulation, and wall thickness were concomitantly reduced, enhancing vessel distensibility.
Cell-autonomous vascular smooth muscle cell PTH1R activity inhibits arteriosclerotic Wnt/beta-catenin signaling and reduces vascular oxidative stress, thus limiting aortic type I collagen and calcium accrual in diabetic LDLR-deficient mice.
血管纤维化和钙化导致糖尿病动脉硬化,损害了为远端组织灌注所必需的血腔生理功能。Wnt 家族成员在“糖脂病”的低水平炎症作用下于动脉中上调,通过β-连环蛋白刺激 I 型胶原表达和间充质祖细胞的成骨矿化。相反,甲状旁腺激素(PTH)抑制低密度脂蛋白受体(LDLR)缺陷型高脂高糖致糖尿病饮食喂养的小鼠的主动脉钙化(HFD)。
我们试图确定血管 PTH 受体(PTH1R)活性对体外和体内动脉粥样硬化性 Wnt/β-连环蛋白信号的影响。我们生成了 SM-caPTH1R 转基因小鼠,这是一种仅在血管中表达组成型激活的 PTH1R 变体 H223R(caPTH1R)的模型。
caPTH1R 抑制了体外的 Wnt/β-连环蛋白信号、胶原蛋白产生以及血管平滑肌细胞增殖和钙化。高脂高糖致糖尿病饮食喂养的转基因 SM-caPTH1R;LDLR(+/-) 小鼠发展为糖脂病,与 LDLR(+/-) 同窝仔相比,空腹血清葡萄糖、胆固醇、体重、体成分或骨量均无改善。SM-caPTH1R 下调主动脉 Col1A1、Runx2 和 Nox1 的表达,而不改变 TNF、Msx2、Wnt7a/b 或 Nox4。SM-caPTH1R 转基因减少了糖尿病 LDLR(+/-) 小鼠主动脉中β-连环蛋白蛋白的积累和信号。主动脉超氧化物(一种激活血管平滑肌细胞中前基质金属蛋白酶 9 和成骨信号的过氧化物前体)水平被 SM-caPTH1R 转基因抑制。主动脉钙化、胶原蛋白积累和壁厚度同时减少,从而提高了血管的伸展性。
细胞自主的血管平滑肌细胞 PTH1R 活性抑制动脉粥样硬化性 Wnt/β-连环蛋白信号并减少血管氧化应激,从而减少糖尿病 LDLR 缺陷型小鼠的主动脉 I 型胶原蛋白和钙积累。