1Department of Human and Clinical Genetics, Leiden University Medical Center, Einthovenweg 20, 2333ZC Leiden, The Netherlands.
Orphanet J Rare Dis. 2010 May 28;5:13. doi: 10.1186/1750-1172-5-13.
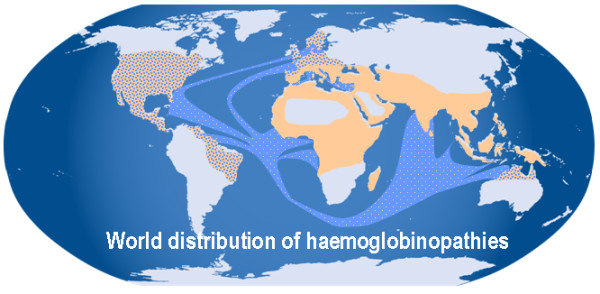
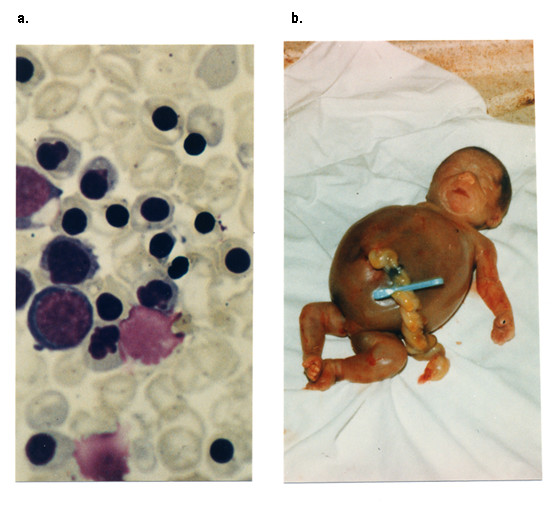
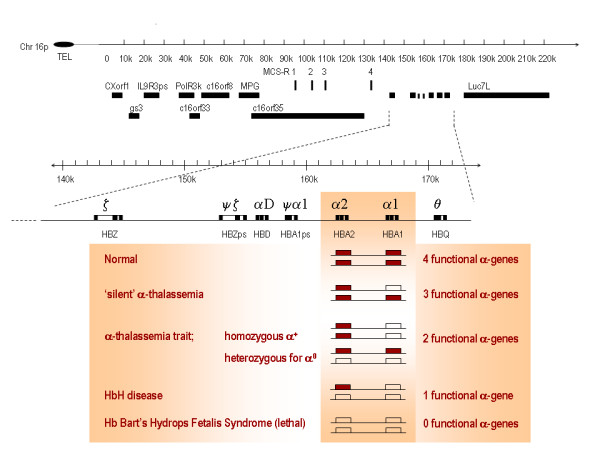
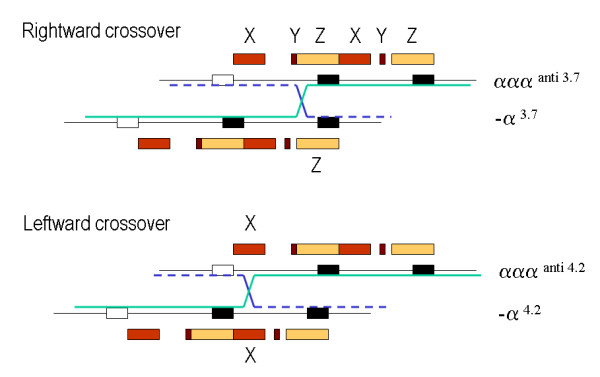
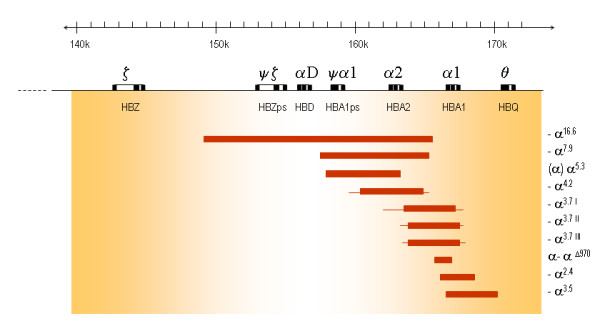
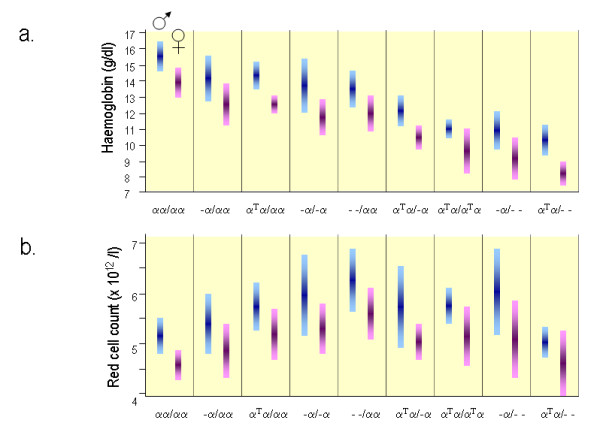
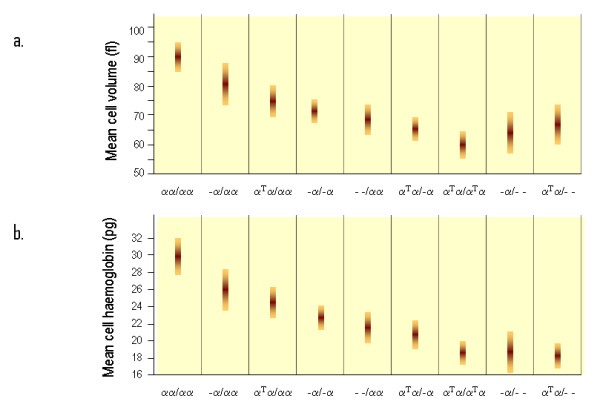
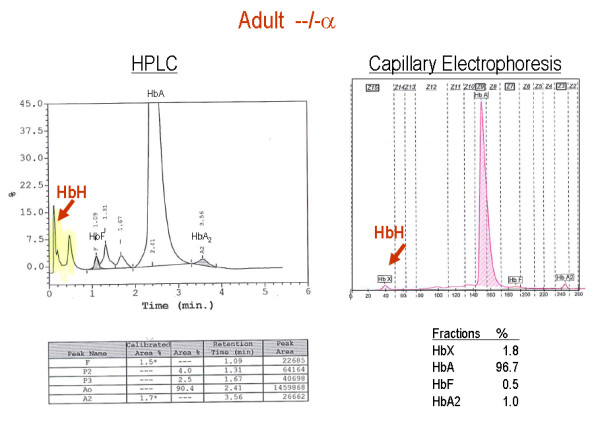
Alpha-thalassaemia is inherited as an autosomal recessive disorder characterised by a microcytic hypochromic anaemia, and a clinical phenotype varying from almost asymptomatic to a lethal haemolytic anaemia.It is probably the most common monogenic gene disorder in the world and is especially frequent in Mediterranean countries, South-East Asia, Africa, the Middle East and in the Indian subcontinent. During the last few decades the incidence of alpha thalassaemia in North-European countries and Northern America has increased because of demographic changes. Compound heterozygotes and some homozygotes have a moderate to severe form of alpha thalassaemia called HbH disease. Hb Bart's hydrops foetalis is a lethal form in which no alpha-globin is synthesized. Alpha thalassaemia most frequently results from deletion of one or both alpha genes from the chromosome and can be classified according to its genotype/phenotype correlation. The normal complement of four functional alpha-globin genes may be decreased by 1, 2, 3 or all 4 copies of the genes, explaining the clinical variation and increasing severity of the disease. All affected individuals have a variable degree of anaemia (low Hb), reduced mean corpuscular haemoglobin (MCH/pg), reduced mean corpuscular volume (MCV/fl) and a normal/slightly reduced level of HbA2. Molecular analysis is usually required to confirm the haematological observations (especially in silent alpha-thalassaemia and alpha-thalassaemia trait). The predominant features in HbH disease are anaemia with variable amounts of HbH (0.8-40%). The type of mutation influences the clinical severity of HbH disease. The distinguishing features of the haemoglobin Bart's hydrops foetalis syndrome are the presence of Hb Bart's and the total absence of HbF. The mode of transmission of alpha thalassaemia is autosomal recessive. Genetic counselling is offered to couples at risk for HbH disease or haemoglobin Bart's Hydrops Foetalis Syndrome. Carriers of alpha+- or alpha0-thalassaemia alleles generally do not need treatment. HbH patients may require intermittent transfusion therapy especially during intercurrent illness. Most pregnancies in which the foetus is known to have the haemoglobin Bart's hydrops foetalis syndrome are terminated due to the increased risk of both maternal and foetal morbidity.
α-地中海贫血是一种常染色体隐性遗传疾病,其特征为小细胞低色素性贫血,临床表现从几乎无症状到致命的溶血性贫血不等。它可能是世界上最常见的单基因遗传病,在地中海国家、东南亚、非洲、中东和印度次大陆尤为常见。在过去几十年中,由于人口结构的变化,北欧国家和北美地区的α-地中海贫血发病率有所增加。复合杂合子和一些纯合子患有称为 HbH 病的中重度α-地中海贫血。HbBart's 胎儿水肿是一种致命形式,其中没有合成α-珠蛋白。α-地中海贫血最常由染色体上一个或两个α 基因缺失引起,可根据其基因型/表型相关性进行分类。正常的四个功能性α-珠蛋白基因的正常拷贝可能减少 1、2、3 或全部 4 个基因,这解释了疾病的临床变异性和严重程度增加。所有受影响的个体都有不同程度的贫血(低 Hb)、平均红细胞血红蛋白量(MCH/pg)降低、平均红细胞体积(MCV/fl)降低和 HbA2 正常/略有降低。通常需要分子分析来确认血液学观察结果(尤其是在沉默性α-地中海贫血和α-地中海贫血特征中)。HbH 病的主要特征是贫血,HbH 含量不定(0.8-40%)。突变类型影响 HbH 病的临床严重程度。HbBart's 胎儿水肿综合征的特征是存在 HbBart's 和完全没有 HbF。α-地中海贫血的遗传方式为常染色体隐性遗传。对 HbH 病或血红蛋白 Bart's 胎儿水肿综合征风险夫妇进行遗传咨询。α+-或α0-地中海贫血等位基因的携带者通常不需要治疗。HbH 患者可能需要间歇性输血治疗,尤其是在并发疾病期间。由于母体和胎儿发病率增加,大多数已知胎儿患有血红蛋白 Bart's 胎儿水肿综合征的妊娠都会终止。