Section of Pediatric Critical Care, Baylor College of Medicine, Houston, TX, USA.
Hepatology. 2010 Jun;51(6):2097-107. doi: 10.1002/hep.23585.
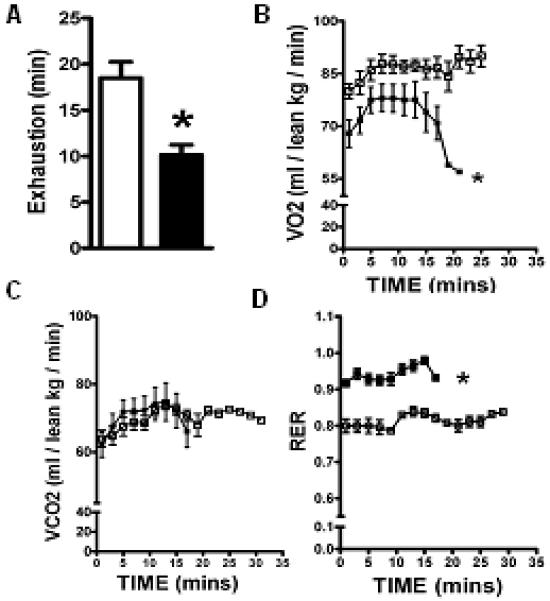
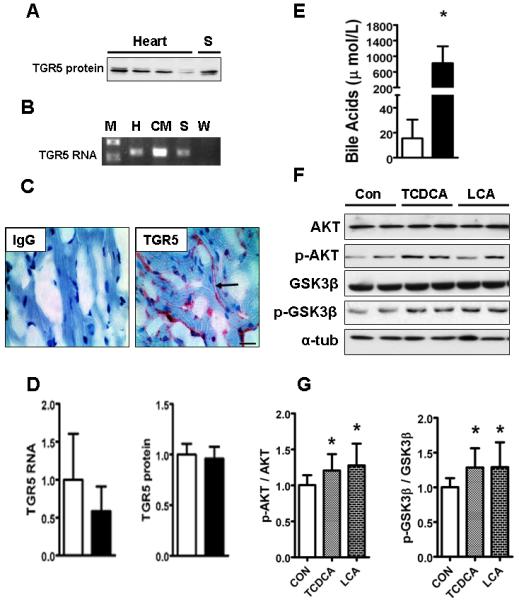
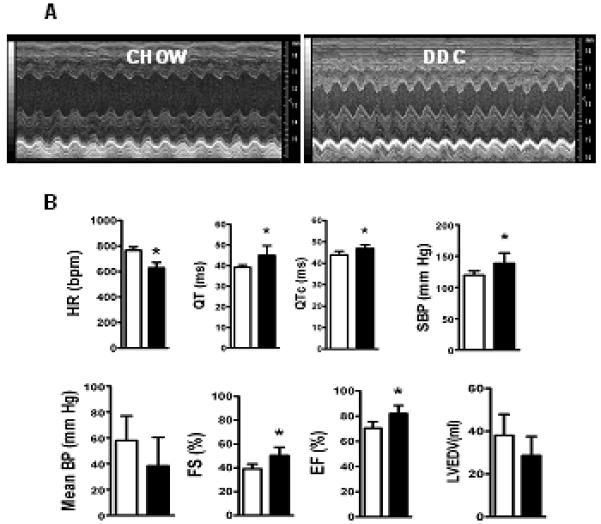
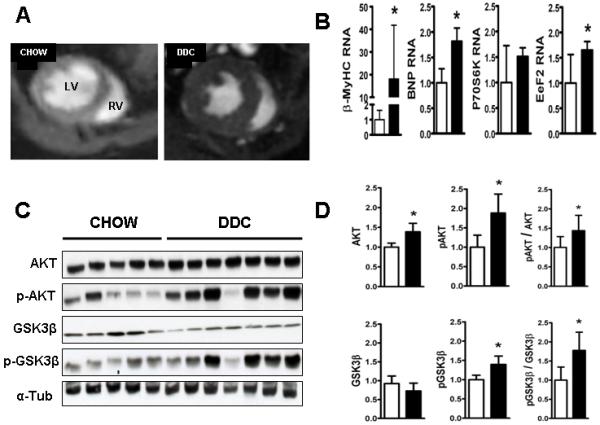
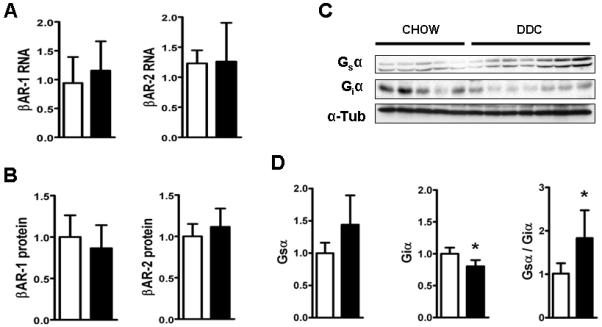
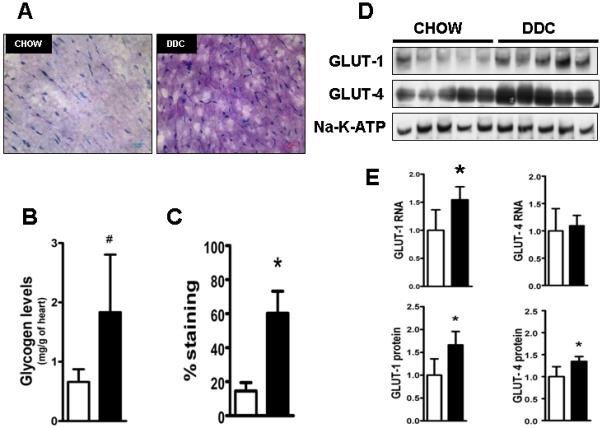
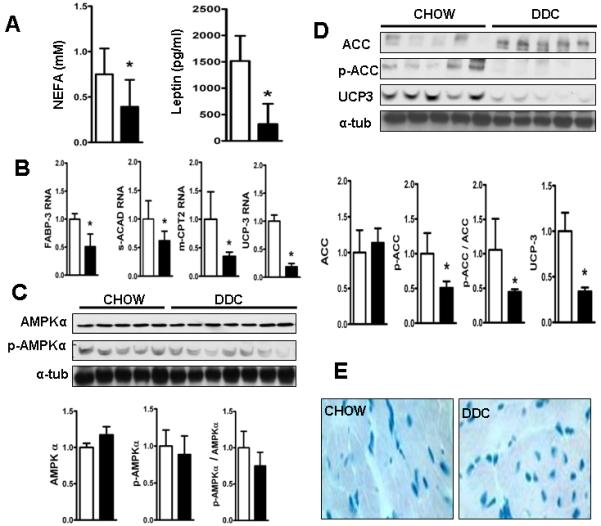
Cardiac dysfunction is a major cause of morbidity and mortality in patients with end-stage liver disease; yet the mechanisms remain largely unknown. We hypothesized that the complex interrelated impairments in cardiac structure and function secondary to progression of liver diseases involve alterations in signaling pathways engaged in cardiac energy metabolism and hypertrophy, augmented by direct effects of high circulating levels of bile acids. Biliary fibrosis was induced in male C57BL/6J mice by feeding a 0.1% 3,5-diethoxycarbonyl-1,4-dihydroxychollidine (DDC) supplemented diet. After 3 weeks, mice underwent live imaging (dual energy x-ray absorptiometry [DEXA] scanning, two-dimensional echocardiography [2DE], electrocardiography, cardiac magnetic resonance imaging), exercise treadmill testing, and histological and biochemical analyses of livers and hearts. Compared with chow-fed mice, DDC-fed mice fatigued earlier on the treadmill, with reduced VO(2). Marked changes were identified electrophysiologically (bradycardia and prolonged QT interval) and functionally (hyperdynamic left ventricular [LV] contractility along with increased LV thickness). Hearts of DDC-fed mice showed hypertrophic signaling (activation of v-akt murine thymoma viral oncogene/protein kinase B [AKT], inhibition of glycogen synthase kinase-3beta [GSK3beta], a 20-fold up-regulation of beta myosin heavy chain RNA and elevated G(s)alpha/G(i)alpha ratio. Genes regulating cardiac fatty acid oxidation pathways were suppressed, along with a threefold increase in myocardial glycogen content. Treatment of mouse cardiomyocytes (which express the membrane bile acid receptor TGR5) with potent natural TGR5 agonists, taurochenodeoxycholic acid and lithocholic acid, activated AKT and inhibited GSK3beta, similar to the changes seen in DDC-fed mouse hearts. This provides support for a novel mechanism whereby circulating natural bile acids can induce signaling pathways in heart associated with hypertrophy.
Three weeks of DDC feeding-induced biliary fibrosis leads to multiple functional, metabolic, electrophysiological, and hypertrophic adaptations in the mouse heart, recapitulating some of the features of human cirrhotic cardiomyopathy.
心脏功能障碍是终末期肝病患者发病率和死亡率的主要原因;然而,其机制在很大程度上尚不清楚。我们假设,由于肝脏疾病的进展导致的心脏结构和功能的复杂相互关联的损伤涉及到心脏能量代谢和肥大相关信号通路的改变,同时也受到循环中高浓度胆汁酸的直接影响。通过给予 0.1%的 3,5-二乙氧羰基-1,4-二羟基胆甾烷(DDC)补充饮食,在雄性 C57BL/6J 小鼠中诱导胆管纤维化。3 周后,对小鼠进行活体成像(双能 X 射线吸收法[DEXA]扫描、二维超声心动图[2DE]、心电图、心脏磁共振成像)、跑步机测试以及肝脏和心脏的组织学和生化分析。与喂食标准饮食的小鼠相比,DDC 喂养的小鼠在跑步机上更早疲劳,VO2 减少。电生理学上有明显的变化(心动过缓和 QT 间期延长)和功能变化(左心室[LV]高动力收缩力增加,同时 LV 厚度增加)。DDC 喂养的小鼠心脏表现出肥大信号(v-akt 鼠胸腺瘤病毒癌基因/蛋白激酶 B [AKT]激活,糖原合成激酶-3beta [GSK3beta]抑制,β肌球蛋白重链 RNA 上调 20 倍,G(s)alpha/G(i)alpha 比值升高)。调节心脏脂肪酸氧化途径的基因受到抑制,心肌糖原含量增加 3 倍。用强的天然 TGR5 激动剂牛磺胆酸和石胆酸处理小鼠心肌细胞(表达膜胆汁酸受体 TGR5),激活 AKT 并抑制 GSK3beta,与 DDC 喂养的小鼠心脏的变化相似。这为一种新的机制提供了支持,即循环中的天然胆汁酸可以诱导与肥大相关的心脏信号通路。
3 周的 DDC 喂养诱导的胆管纤维化导致小鼠心脏出现多种功能、代谢、电生理和肥大适应性,再现了人类肝硬化心肌病的一些特征。