Department of Computer Science & Engineering, The Chinese University of Hong Kong, and Hong Kong Bioinformatics Centre, Shatin, NT, Hong Kong, China.
Nucleic Acids Res. 2010 Oct;38(19):6324-37. doi: 10.1093/nar/gkq500. Epub 2010 Jun 6.
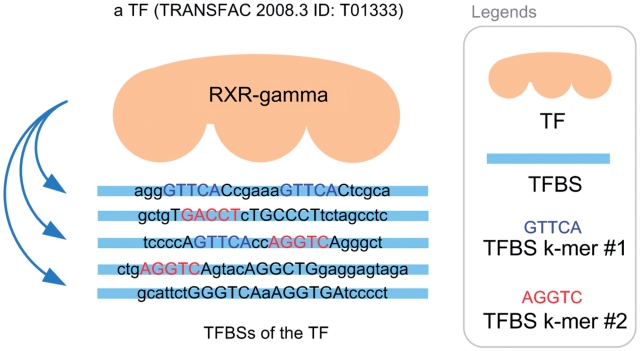
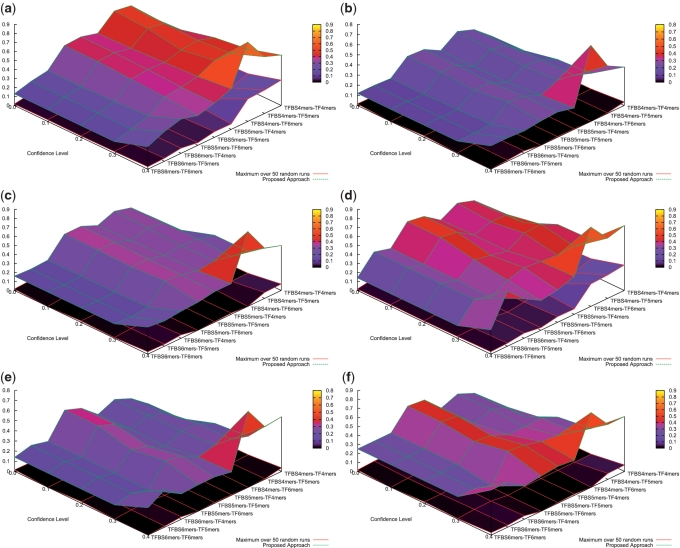
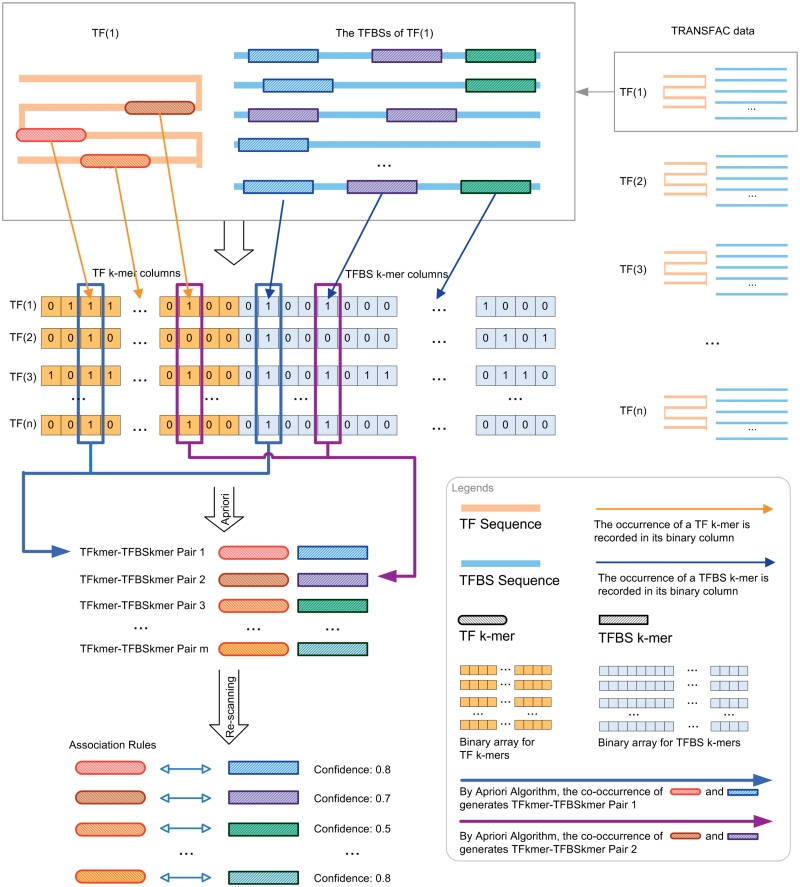
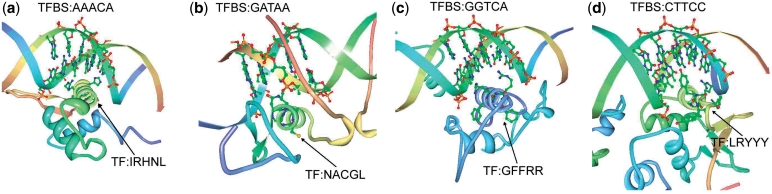
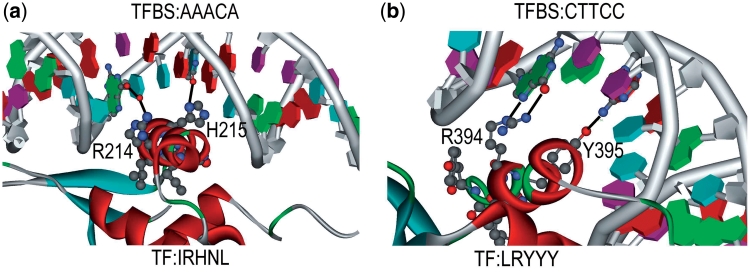
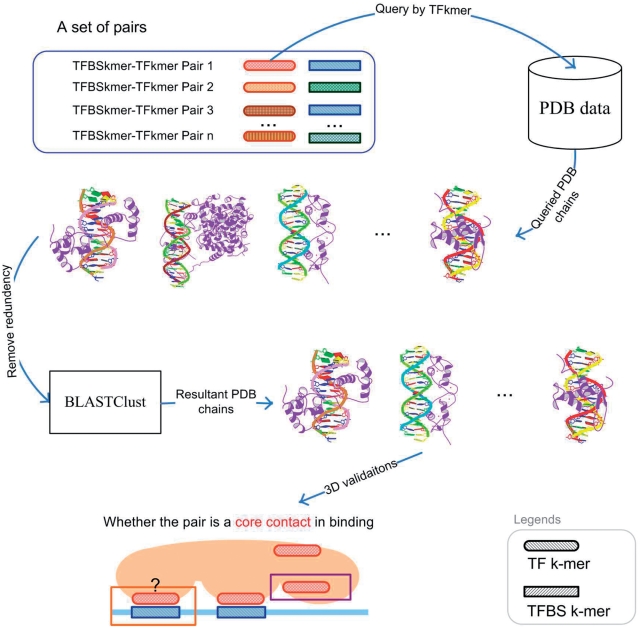
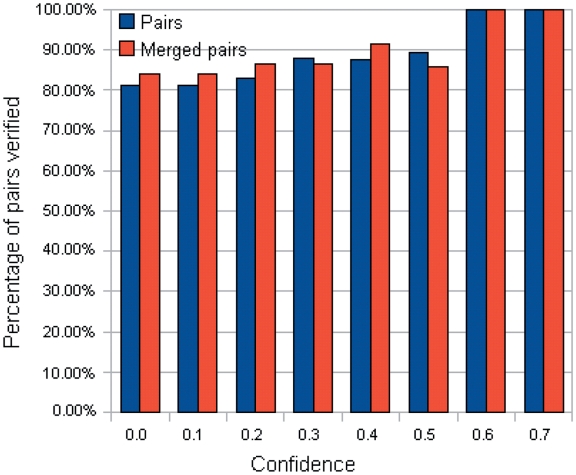
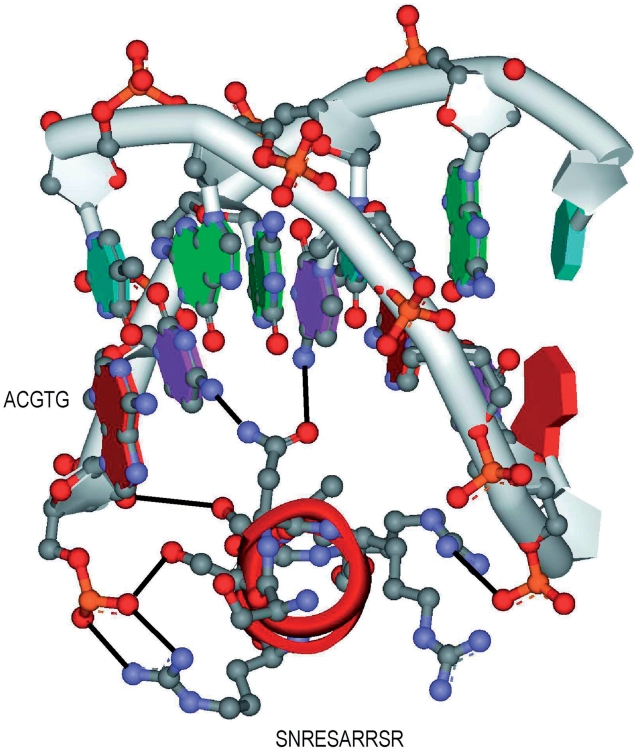
Protein-DNA bindings between transcription factors (TFs) and transcription factor binding sites (TFBSs) play an essential role in transcriptional regulation. Over the past decades, significant efforts have been made to study the principles for protein-DNA bindings. However, it is considered that there are no simple one-to-one rules between amino acids and nucleotides. Many methods impose complicated features beyond sequence patterns. Protein-DNA bindings are formed from associated amino acid and nucleotide sequence pairs, which determine many functional characteristics. Therefore, it is desirable to investigate associated sequence patterns between TFs and TFBSs. With increasing computational power, availability of massive experimental databases on DNA and proteins, and mature data mining techniques, we propose a framework to discover associated TF-TFBS binding sequence patterns in the most explicit and interpretable form from TRANSFAC. The framework is based on association rule mining with Apriori algorithm. The patterns found are evaluated by quantitative measurements at several levels on TRANSFAC. With further independent verifications from literatures, Protein Data Bank and homology modeling, there are strong evidences that the patterns discovered reveal real TF-TFBS bindings across different TFs and TFBSs, which can drive for further knowledge to better understand TF-TFBS bindings.
转录因子(TFs)与转录因子结合位点(TFBSs)之间的蛋白-DNA 结合对于转录调控起着至关重要的作用。在过去的几十年中,人们已经做出了巨大的努力来研究蛋白-DNA 结合的原理。然而,人们认为氨基酸和核苷酸之间没有简单的一一对应规则。许多方法引入了复杂的特征,而不仅仅是序列模式。蛋白-DNA 结合是由相关的氨基酸和核苷酸序列对形成的,这些序列对决定了许多功能特征。因此,研究 TF 和 TFBS 之间的相关序列模式是很有必要的。随着计算能力的提高、大量 DNA 和蛋白质实验数据库的可用性以及成熟的数据挖掘技术的出现,我们提出了一个从 TRANSFAC 中以最明确和可解释的形式发现相关 TF-TFBS 结合序列模式的框架。该框架基于关联规则挖掘和 Apriori 算法。所发现的模式通过在 TRANSFAC 上进行几个层次的定量测量进行评估。通过进一步从文献、蛋白质数据库和同源建模中进行独立验证,有强有力的证据表明,所发现的模式揭示了不同 TF 和 TFBS 之间的真实 TF-TFBS 结合,这可以进一步深入了解 TF-TFBS 结合。