Delft Bioinformatics Lab, Delft University of Technology, 2628 CD Delft, The Netherlands.
Bioinformatics. 2010 Jun 15;26(12):i149-57. doi: 10.1093/bioinformatics/btq211.
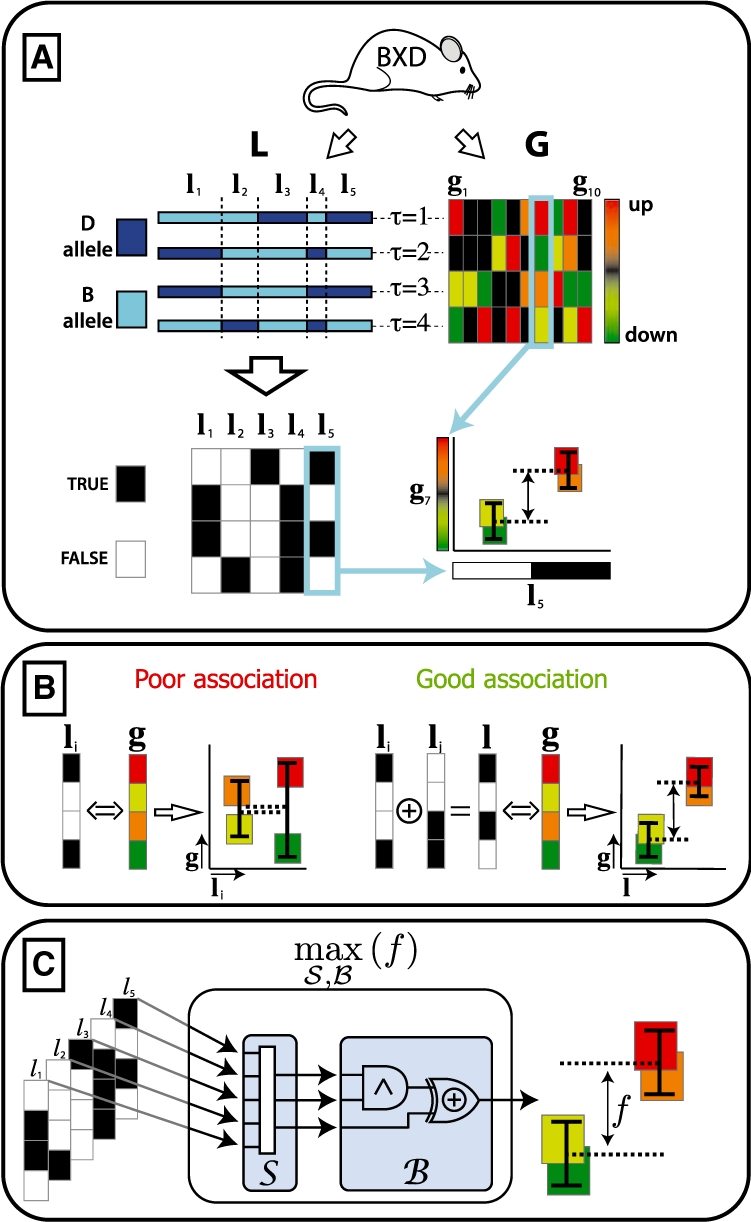
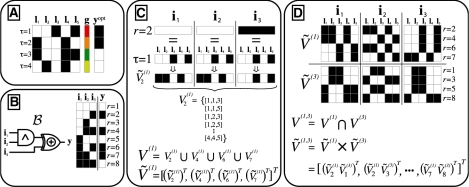
We propose an efficient method to infer combinatorial association logic networks from multiple genome-wide measurements from the same sample. We demonstrate our method on a genetical genomics dataset, in which we search for Boolean combinations of multiple genetic loci that associate with transcript levels.
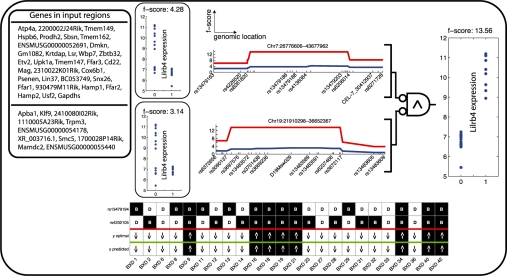
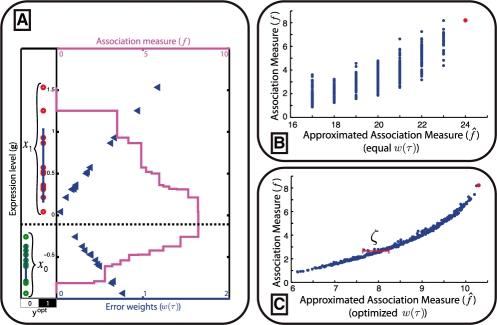
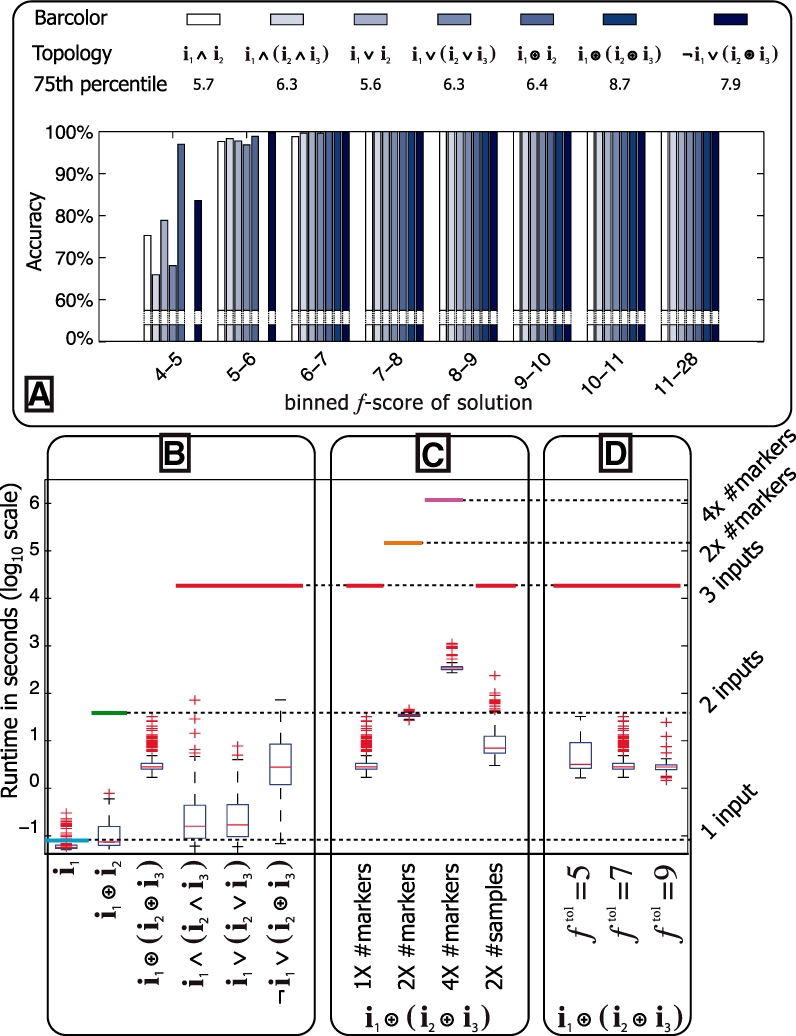
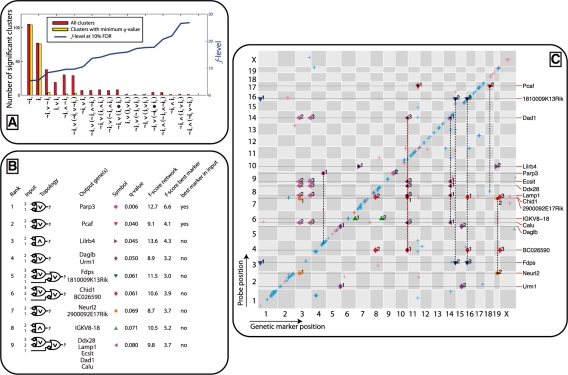
Our method provably finds the global solution and is very efficient with runtimes of up to four orders of magnitude faster than the exhaustive search. This enables permutation procedures for determining accurate false positive rates and allows selection of the most parsimonious model. When applied to transcript levels measured in myeloid cells from 24 genotyped recombinant inbred mouse strains, we discovered that nine gene clusters are putatively modulated by a logical combination of trait loci rather than a single locus. A literature survey supports and further elucidates one of these findings. Due to our approach, optimal solutions for multi-locus logic models and accurate estimates of the associated false discovery rates become feasible. Our algorithm, therefore, offers a valuable alternative to approaches employing complex, albeit suboptimal optimization strategies to identify complex models.
The MATLAB code of the prototype implementation is available on: http://bioinformatics.tudelft.nl/ or http://bioinformatics.nki.nl/.
我们提出了一种从同一个样本的多个全基因组测量中推断组合关联逻辑网络的有效方法。我们在一个遗传基因组学数据集上展示了我们的方法,在该数据集中,我们搜索与转录水平相关的多个遗传位点的布尔组合。
我们的方法可以证明找到全局解,并且非常高效,运行时间比穷举搜索快高达四个数量级。这使得可以进行置换程序以确定准确的假阳性率,并允许选择最简约的模型。当应用于从 24 个基因分型重组近交系小鼠骨髓细胞中测量的转录水平时,我们发现九个基因簇可能是由特征位点的逻辑组合而不是单个位点调节的。文献综述支持并进一步阐明了其中一个发现。由于我们的方法,多基因座逻辑模型的最优解决方案和相关假发现率的准确估计成为可能。因此,我们的算法为使用复杂但非最优优化策略来识别复杂模型的方法提供了一种有价值的替代方法。
原型实现的 MATLAB 代码可在以下网址获得:http://bioinformatics.tudelft.nl/ 或 http://bioinformatics.nki.nl/。