Department of Computer Science, University of California Los Angeles, Los Angeles, CA 90095, USA.
Bioinformatics. 2010 Jun 15;26(12):i183-90. doi: 10.1093/bioinformatics/btq215.
Haplotype inference is an important step for many types of analyses of genetic variation in the human genome. Traditional approaches for obtaining haplotypes involve collecting genotype information from a population of individuals and then applying a haplotype inference algorithm. The development of high-throughput sequencing technologies allows for an alternative strategy to obtain haplotypes by combining sequence fragments. The problem of 'haplotype assembly' is the problem of assembling the two haplotypes for a chromosome given the collection of such fragments, or reads, and their locations in the haplotypes, which are pre-determined by mapping the reads to a reference genome. Errors in reads significantly increase the difficulty of the problem and it has been shown that the problem is NP-hard even for reads of length 2. Existing greedy and stochastic algorithms are not guaranteed to find the optimal solutions for the haplotype assembly problem.
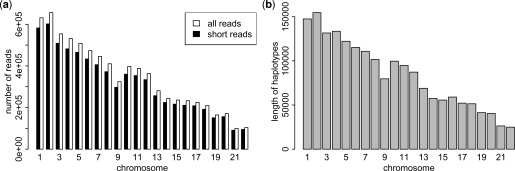
In this article, we proposed a dynamic programming algorithm that is able to assemble the haplotypes optimally with time complexity O(m x 2(k) x n), where m is the number of reads, k is the length of the longest read and n is the total number of SNPs in the haplotypes. We also reduce the haplotype assembly problem into the maximum satisfiability problem that can often be solved optimally even when k is large. Taking advantage of the efficiency of our algorithm, we perform simulation experiments demonstrating that the assembly of haplotypes using reads of length typical of the current sequencing technologies is not practical. However, we demonstrate that the combination of this approach and the traditional haplotype phasing approaches allow us to practically construct haplotypes containing both common and rare variants.
单倍型推断是人类基因组中遗传变异分析的许多类型的重要步骤。传统的方法来获得单倍型涉及从个体的人群中收集基因型信息,然后应用单倍型推断算法。高通量测序技术的发展允许通过组合序列片段获得单倍型的替代策略。“单倍型组装”问题是给定此类片段(或读取)的集合以及它们在由将读取映射到参考基因组而预先确定的单倍型中的位置,组装染色体的两个单倍型的问题。读取中的错误大大增加了问题的难度,并且已经表明,即使对于长度为 2 的读取,该问题也是 NP 难的。现有的贪婪和随机算法不能保证找到单倍型组装问题的最优解。
在本文中,我们提出了一种动态规划算法,该算法能够以时间复杂度 O(m x 2(k) x n)最优地组装单倍型,其中 m 是读取的数量,k 是最长读取的长度,n 是单倍型中 SNP 的总数。我们还将单倍型组装问题简化为最大可满足性问题,即使 k 很大,通常也可以最优地解决该问题。利用我们算法的效率,我们进行了模拟实验,证明使用当前测序技术的典型长度的读取进行单倍型组装是不切实际的。但是,我们证明了这种方法与传统的单倍型相位方法相结合,使我们能够实际构建包含常见和罕见变体的单倍型。