Evolutionary Systems Biology Group, SRI International, Menlo Park, CA, USA.
BMC Bioinformatics. 2010 Jun 9;11:312. doi: 10.1186/1471-2105-11-312.
Phylogenetic relationships between genes are not only of theoretical interest: they enable us to learn about human genes through the experimental work on their relatives in numerous model organisms from bacteria to fruit flies and mice. Yet the most commonly used computational algorithms for reconstructing gene trees can be inaccurate for numerous reasons, both algorithmic and biological. Additional information beyond gene sequence data has been shown to improve the accuracy of reconstructions, though at great computational cost.
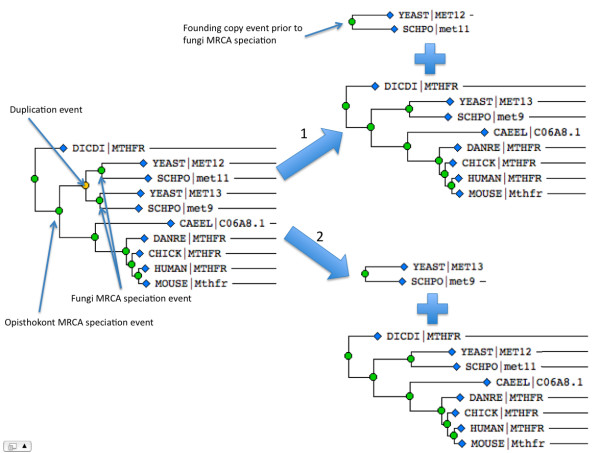
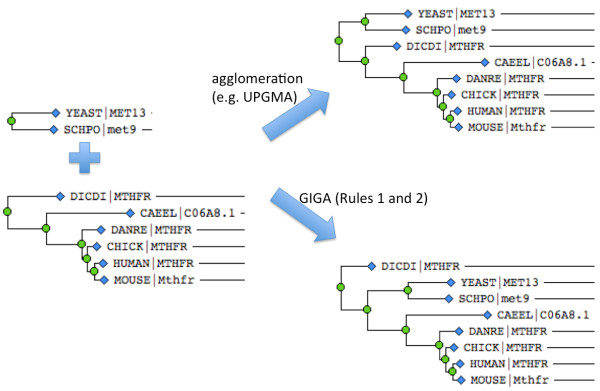
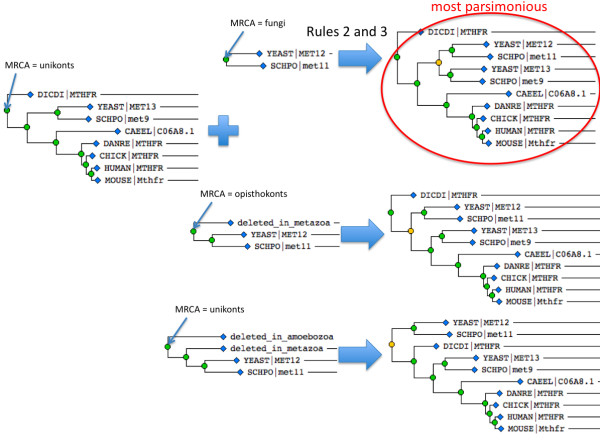
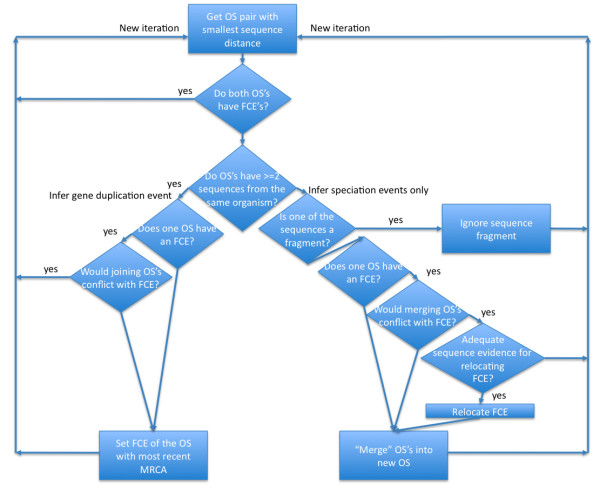
We describe a simple, fast algorithm for inferring gene phylogenies, which makes use of information that was not available prior to the genomic age: namely, a reliable species tree spanning much of the tree of life, and knowledge of the complete complement of genes in a species' genome. The algorithm, called GIGA, constructs trees agglomeratively from a distance matrix representation of sequences, using simple rules to incorporate this genomic age information. GIGA makes use of a novel conceptualization of gene trees as being composed of orthologous subtrees (containing only speciation events), which are joined by other evolutionary events such as gene duplication or horizontal gene transfer. An important innovation in GIGA is that, at every step in the agglomeration process, the tree is interpreted/reinterpreted in terms of the evolutionary events that created it. Remarkably, GIGA performs well even when using a very simple distance metric (pairwise sequence differences) and no distance averaging over clades during the tree construction process.
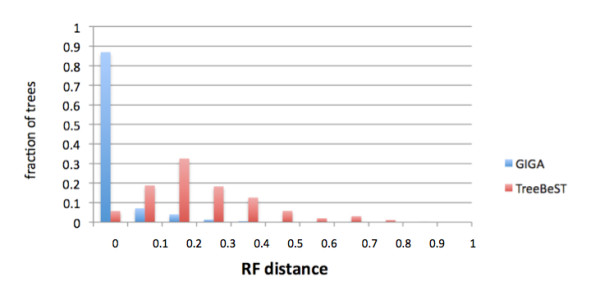
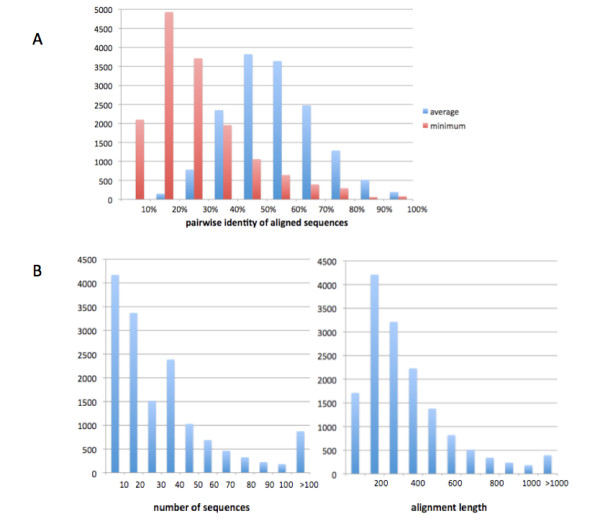
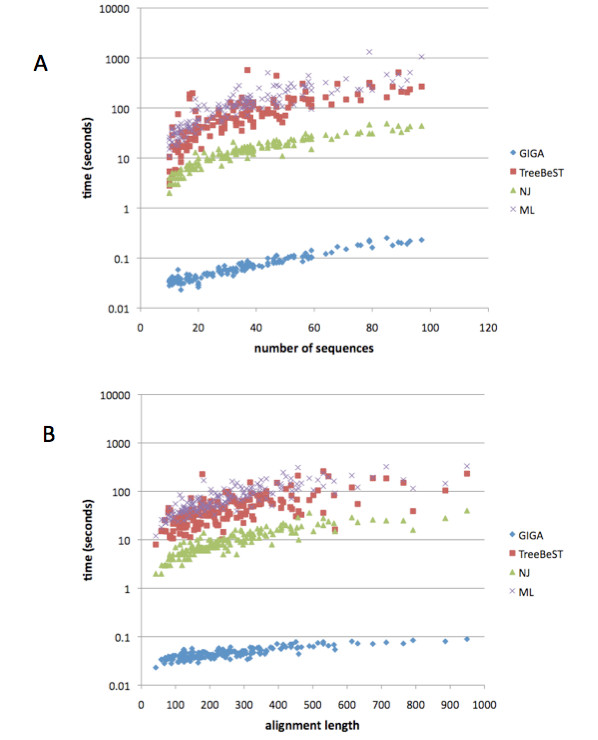
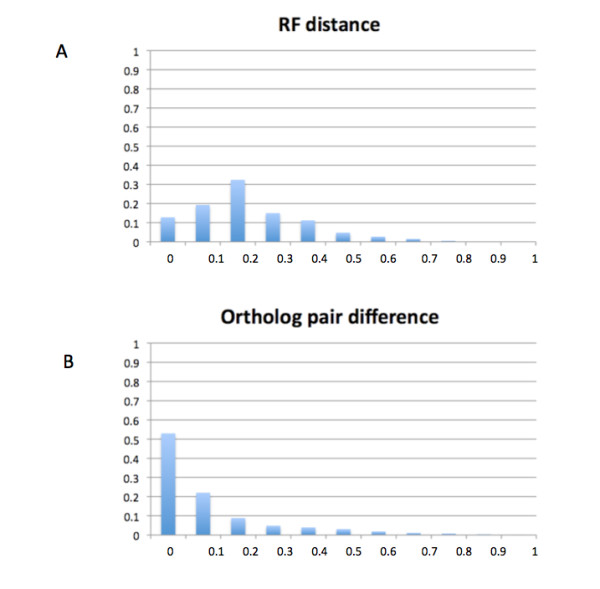
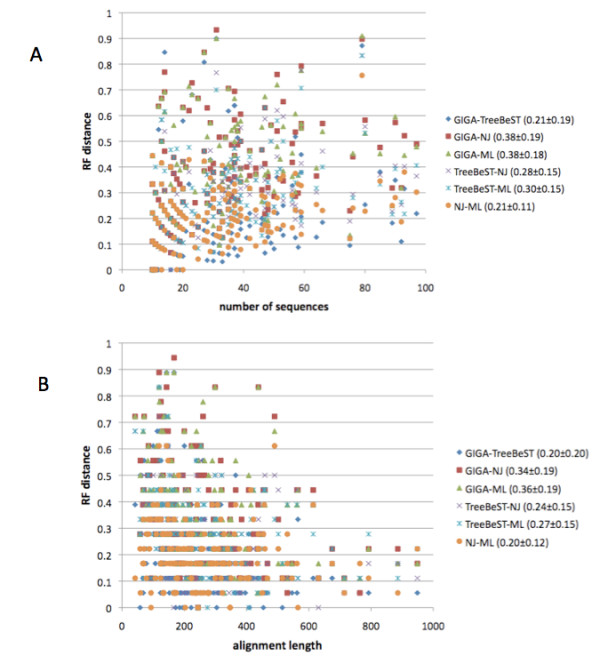
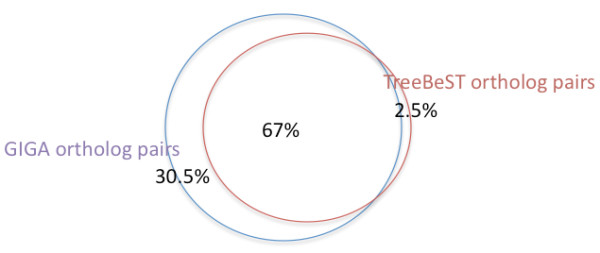
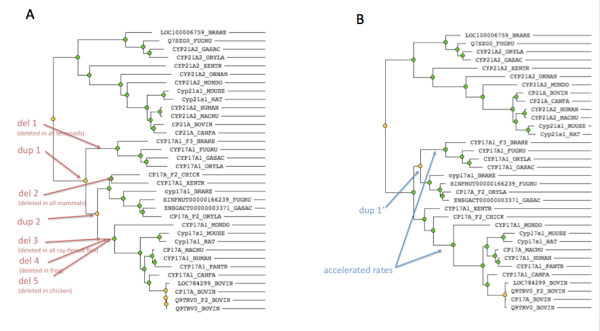
GIGA is efficient, allowing phylogenetic reconstruction of very large gene families and determination of orthologs on a large scale. It is exceptionally robust to adding more gene sequences, opening up the possibility of creating stable identifiers for referring to not only extant genes, but also their common ancestors. We compared trees produced by GIGA to those in the TreeFam database, and they were very similar in general, with most differences likely due to poor alignment quality. However, some remaining differences are algorithmic, and can be explained by the fact that GIGA tends to put a larger emphasis on minimizing gene duplication and deletion events.
基因之间的系统发育关系不仅具有理论意义:通过在从细菌到果蝇和老鼠的众多模式生物中对其亲缘基因的实验工作,我们可以了解人类基因。然而,由于算法和生物学方面的诸多原因,最常用的用于重建基因树的计算算法可能不准确。除了基因序列数据之外,额外的信息已被证明可以提高重建的准确性,尽管计算成本很高。
我们描述了一种简单、快速的推断基因系统发育的算法,该算法利用了基因组时代之前不可用的信息:即跨越生命之树的可靠物种树,以及物种基因组中完整基因组成的知识。该算法称为 GIGA,它使用序列的距离矩阵表示来进行聚类,使用简单的规则来合并这种基因组时代的信息。GIGA 将基因树概念化为由同源子树(仅包含物种形成事件)组成,通过其他进化事件(如基因复制或水平基因转移)将它们连接在一起。GIGA 的一个重要创新是,在聚类过程的每一步,都根据创建它的进化事件来解释/重新解释树。值得注意的是,即使使用非常简单的距离度量(成对序列差异)并且在树构建过程中不对类群进行距离平均,GIGA 的性能也很好。
GIGA 效率高,允许对非常大的基因家族进行系统发育重建,并大规模确定同源基因。它非常稳健,可以添加更多基因序列,不仅为现存基因,而且为其共同祖先创建稳定的标识符。我们将 GIGA 生成的树与 TreeFam 数据库中的树进行了比较,它们总体上非常相似,大多数差异可能是由于对齐质量差造成的。但是,一些剩余的差异是算法性的,可以通过 GIGA 倾向于更强调最小化基因复制和缺失事件来解释。