Institute of Child Health, University College London with Great Ormond Street Hospital for Children, National Health Service Trust, London, UK.
Brain. 2010 Jul;133(Pt 7):2148-59. doi: 10.1093/brain/awq143. Epub 2010 Jun 16.
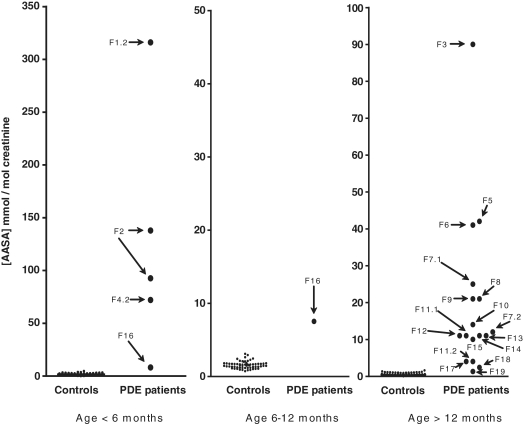
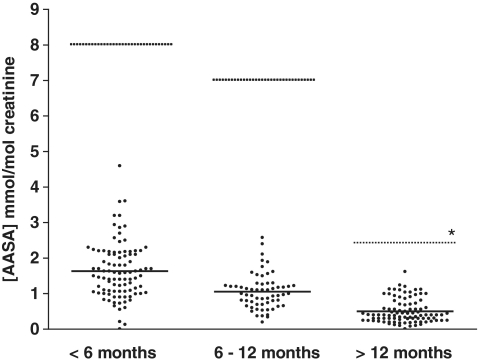
Pyridoxine-dependent epilepsy was recently shown to be due to mutations in the ALDH7A1 gene, which encodes antiquitin, an enzyme that catalyses the nicotinamide adenine dinucleotide-dependent dehydrogenation of l-alpha-aminoadipic semialdehyde/L-Delta1-piperideine 6-carboxylate. However, whilst this is a highly treatable disorder, there is general uncertainty about when to consider this diagnosis and how to test for it. This study aimed to evaluate the use of measurement of urine L-alpha-aminoadipic semialdehyde/creatinine ratio and mutation analysis of ALDH7A1 (antiquitin) in investigation of patients with suspected or clinically proven pyridoxine-dependent epilepsy and to characterize further the phenotypic spectrum of antiquitin deficiency. Urinary L-alpha-aminoadipic semialdehyde concentration was determined by liquid chromatography tandem mass spectrometry. When this was above the normal range, DNA sequencing of the ALDH7A1 gene was performed. Clinicians were asked to complete questionnaires on clinical, biochemical, magnetic resonance imaging and electroencephalography features of patients. The clinical spectrum of antiquitin deficiency extended from ventriculomegaly detected on foetal ultrasound, through abnormal foetal movements and a multisystem neonatal disorder, to the onset of seizures and autistic features after the first year of life. Our relatively large series suggested that clinical diagnosis of pyridoxine dependent epilepsy can be challenging because: (i) there may be some response to antiepileptic drugs; (ii) in infants with multisystem pathology, the response to pyridoxine may not be instant and obvious; and (iii) structural brain abnormalities may co-exist and be considered sufficient cause of epilepsy, whereas the fits may be a consequence of antiquitin deficiency and are then responsive to pyridoxine. These findings support the use of biochemical and DNA tests for antiquitin deficiency and a clinical trial of pyridoxine in infants and children with epilepsy across a broad range of clinical scenarios.
吡哆醇依赖性癫痫最近被证明是由于 ALDH7A1 基因突变引起的,该基因突变导致抗衰素(antiquitin)的缺失,抗衰素是一种酶,能够催化烟酰胺腺嘌呤二核苷酸依赖性 l-α-氨基己二酸半醛/L-Δ1-哌啶-6-羧酸的脱氢反应。然而,尽管这是一种高度可治疗的疾病,但对于何时考虑这种诊断以及如何进行检测,普遍存在不确定性。本研究旨在评估尿液 l-α-氨基己二酸半醛/肌酐比值的测量和 ALDH7A1(抗衰素)突变分析在疑似或临床确诊的吡哆醇依赖性癫痫患者中的应用,并进一步阐明抗衰素缺乏症的表型谱。通过液相色谱串联质谱法测定尿液 l-α-氨基己二酸半醛浓度。当该浓度超过正常范围时,进行 ALDH7A1 基因测序。临床医生被要求填写有关患者临床、生化、磁共振成像和脑电图特征的问卷。抗衰素缺乏症的临床谱从胎儿超声检查发现的脑室扩大,到异常胎儿运动和多系统新生儿疾病,再到发病后第一年的癫痫发作和自闭症特征,范围广泛。我们相对较大的系列研究表明,吡哆醇依赖性癫痫的临床诊断可能具有挑战性,原因如下:(i)可能对抗癫痫药物有一定的反应;(ii)在多系统病理的婴儿中,对吡哆醇的反应可能不会立即明显;(iii)结构脑异常可能同时存在,并被认为是癫痫的充分原因,而癫痫发作可能是抗衰素缺乏的结果,然后对吡哆醇有反应。这些发现支持对抗衰素缺乏症进行生化和 DNA 检测,并在广泛的临床情况下对患有癫痫的婴儿和儿童进行吡哆醇临床试验。