Department of Microbiology, University of Illinois at Urbana-Champaign.
Mol Biol Evol. 2011 Jan;28(1):211-21. doi: 10.1093/molbev/msq185. Epub 2010 Aug 2.
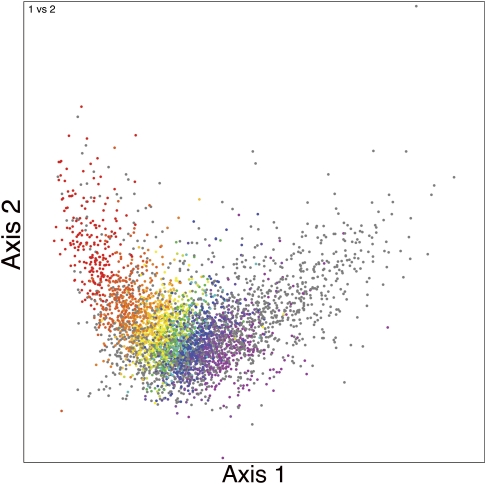
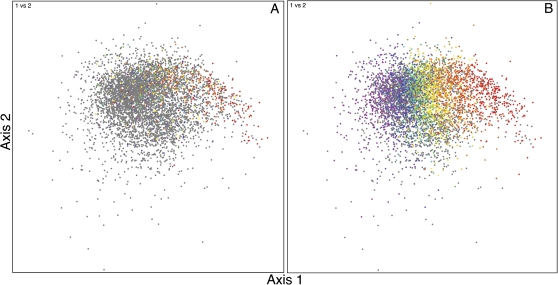
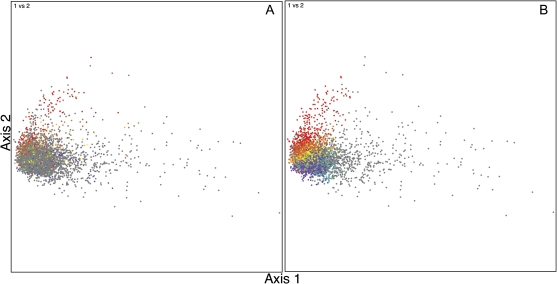
Codon usage can provide insights into the nature of the genes in a genome. Genes that are "native" to a genome (have not been recently acquired by horizontal transfer) range in codon usage from a low-bias "typical" usage to a more biased "high-expression" usage characteristic of genes encoding abundant proteins. Genes that differ from these native codon usages are candidates for foreign genes that have been recently acquired by horizontal gene transfer. In this study, we present a method for characterizing the codon usages of native genes--both typical and highly expressed--within a genome. Each gene is evaluated relative to a half line (or axis) in a 59D space of codon usage. The axis begins at the modal codon usage, the usage that matches the largest number of genes in the genome, and it passes through a point representing the codon usage of a set of genes with expression-related bias. A gene whose codon usage matches (does not significantly differ from) a point on this axis is a candidate native gene, and the location of its projection onto the axis provides a general estimate of its expression level. A gene that differs significantly from all points on the axis is a candidate foreign gene. This automated approach offers significant improvements over existing methods. We illustrate this by analyzing the genomes of Pseudomonas aeruginosa PAO1 and Bacillus anthracis A0248, which can be difficult to analyze with commonly used methods due to their biased base compositions. Finally, we use this approach to measure the proportion of candidate foreign genes in 923 bacterial and archaeal genomes. The organisms with the most homogeneous genomes (containing the fewest candidate foreign genes) are mostly endosymbionts and parasites, though with exceptions that include Pelagibacter ubique and Beutenbergia cavernae. The organisms with the most heterogeneous genomes (containing the most candidate foreign genes) include members of the genera Bacteroides, Corynebacterium, Desulfotalea, Neisseria, Xylella, and Thermobaculum.
密码子使用情况可以提供有关基因组中基因性质的信息。“本地”(最近未通过水平转移获得)基因的密码子使用范围从低偏向“典型”使用到更偏向“高表达”使用,这是编码丰富蛋白质的基因的特征。与这些本地密码子用法不同的基因是最近通过水平基因转移获得的外来基因的候选者。在这项研究中,我们提出了一种描述基因组中本地基因(包括典型和高度表达的基因)的密码子用法的方法。每个基因相对于密码子用法的 59D 空间中的半线(或轴)进行评估。该轴始于模式密码子用法,即与基因组中大多数基因匹配的用法,并且它通过代表具有表达相关偏向的一组基因的密码子用法的点。其密码子用法与该轴上的点匹配(没有显著差异)的基因是候选本地基因,并且其在轴上的投影的位置提供了其表达水平的一般估计。与轴上的所有点都显著不同的基因是候选外来基因。与现有方法相比,这种自动化方法具有显著的改进。我们通过分析铜绿假单胞菌 PAO1 和炭疽杆菌 A0248 的基因组来说明这一点,由于其偏倚的碱基组成,这些方法通常难以分析。最后,我们使用这种方法来测量 923 个细菌和古细菌基因组中候选外来基因的比例。具有最均匀基因组(包含最少候选外来基因)的生物体主要是内共生体和寄生虫,尽管也有例外,包括 Pelagibacter ubique 和 Beutenbergia cavernae。具有最不均匀基因组(包含最多候选外来基因)的生物体包括拟杆菌属、棒状杆菌属、脱硫脱氮杆菌属、奈瑟菌属、木杆菌属和 Thermobaculum 属的成员。