Kloster Morten, Tang Chao
Department of Bioengineering and Therapeutic Sciences, UCSF, San Francisco, California 94158, USA.
Nucleic Acids Res. 2008 Jun;36(11):3819-27. doi: 10.1093/nar/gkn288. Epub 2008 May 21.
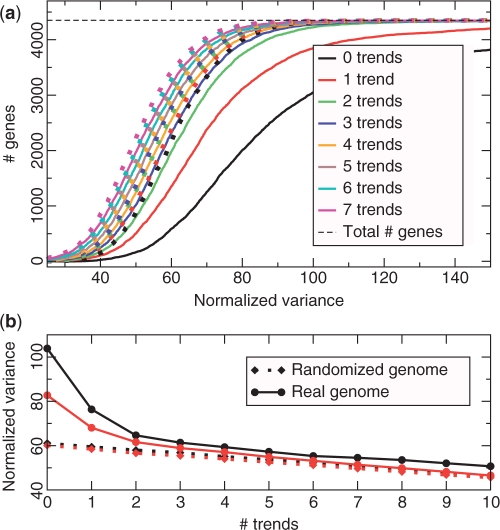
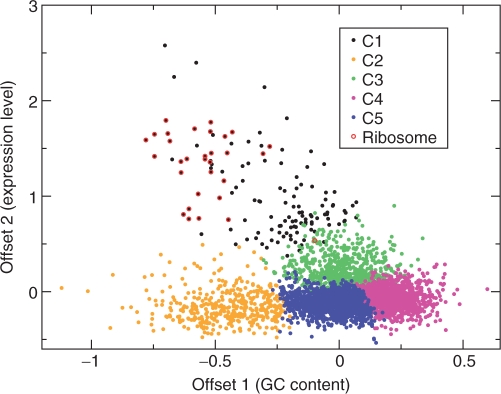
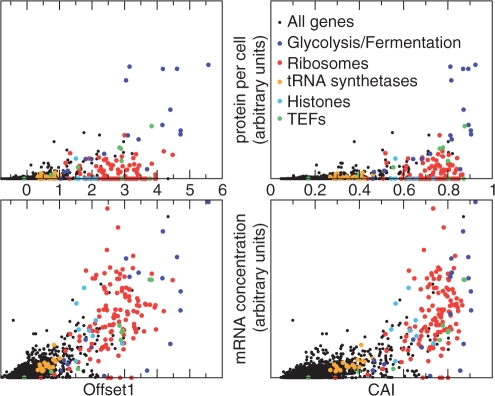
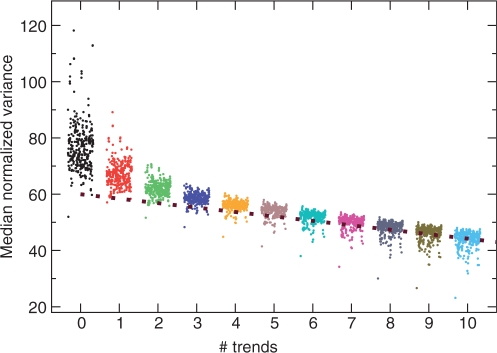
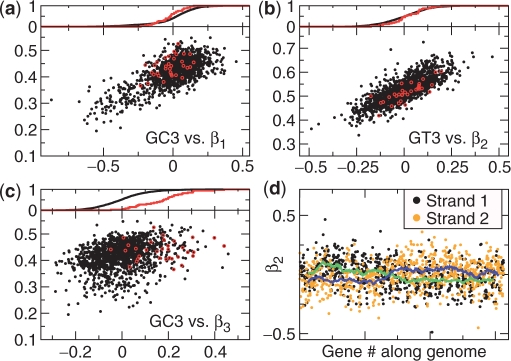
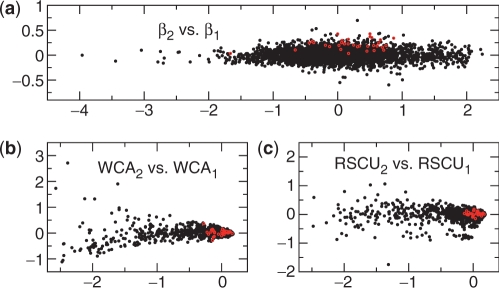
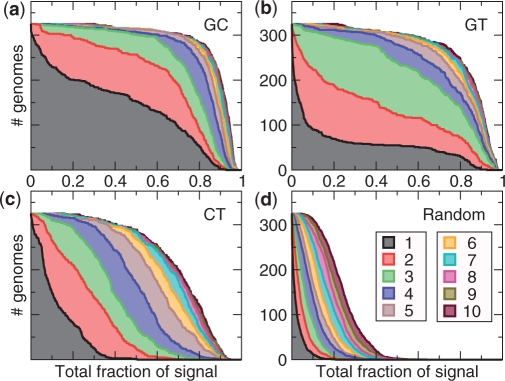
The genetic code is degenerate--most amino acids can be encoded by from two to as many as six different codons. The synonymous codons are not used with equal frequency: not only are some codons favored over others, but also their usage can vary significantly from species to species and between different genes in the same organism. Known causes of codon bias include differences in mutation rates as well as selection pressure related to the expression level of a gene, but the standard analysis methods can account for only a fraction of the observed codon usage variation. We here introduce an explicit model of codon usage bias, inspired by statistical physics. Combining this model with a maximum likelihood approach, we are able to clearly identify different sources of bias in various genomes. We have applied the algorithm to Saccharomyces cerevisiae as well as 325 prokaryote genomes, and in most cases our model explains essentially all observed variance.
遗传密码是简并的——大多数氨基酸可以由2种至多达6种不同的密码子编码。同义密码子的使用频率并不相同:不仅某些密码子比其他密码子更受青睐,而且它们的使用在不同物种之间以及同一生物体的不同基因之间也可能有显著差异。已知密码子偏好的成因包括突变率的差异以及与基因表达水平相关的选择压力,但标准分析方法只能解释所观察到的密码子使用变异的一部分。我们在此引入一个受统计物理学启发的密码子使用偏好显式模型。将此模型与最大似然法相结合,我们能够清晰地识别各种基因组中不同的偏好来源。我们已将该算法应用于酿酒酵母以及325个原核生物基因组,在大多数情况下,我们的模型基本上解释了所有观察到的变异。