Chiò Adriano, Calvo Andrea, Moglia Cristina, Restagno Gabriella, Ossola Irene, Brunetti Maura, Montuschi Anna, Cistaro Angelina, Ticca Anna, Traynor Bryan J, Schymick Jennifer C, Mutani Roberto, Marrosu Maria Giovanna, Murru Maria Rita, Borghero Giuseppe
Department of Neuroscience, University of Turin, via Cherasco 15, 10126 Turin, Italy.
Arch Neurol. 2010 Aug;67(8):1002-9. doi: 10.1001/archneurol.2010.173.
TAR DNA-binding protein 43, encoded by the TARDBP gene, has been identified as the major pathological protein of frontotemporal lobar dementia (FTLD) with or without amyotrophic lateral sclerosis (ALS) and sporadic ALS. Subsequently, mutations in the TARDBP gene have been detected in 2% to 3% of patients with ALS (both familial and sporadic ALS). However, to our knowledge, there is only 1 description of 2 patients with FTLD and TARDBP gene mutations who later developed motor neuron disease.
To describe cognitive abnormalities in 3 Italian families with familial ALS and TARDBP gene mutations.
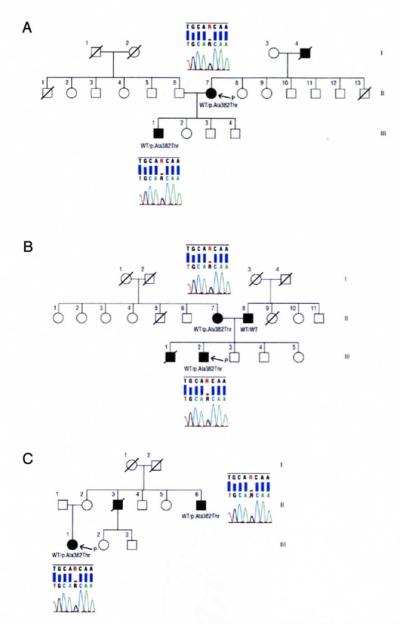
DESIGN, SETTING, AND PARTICIPANTS: Genetic, neuropsychological, and neuroimaging analyses in 36 patients with familial non-superoxide dismutase 1 gene (SOD1) ALS and 280 healthy controls. Main Outcome Measure We identified 3 index cases of familial ALS carrying the p.Ala382Thr missense mutation of the TARDBP gene and with clinical, neuroimaging, and neuropsychological features of FTLD.
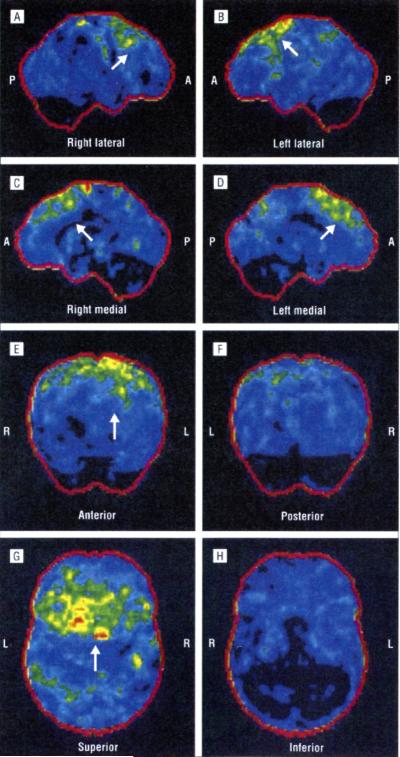
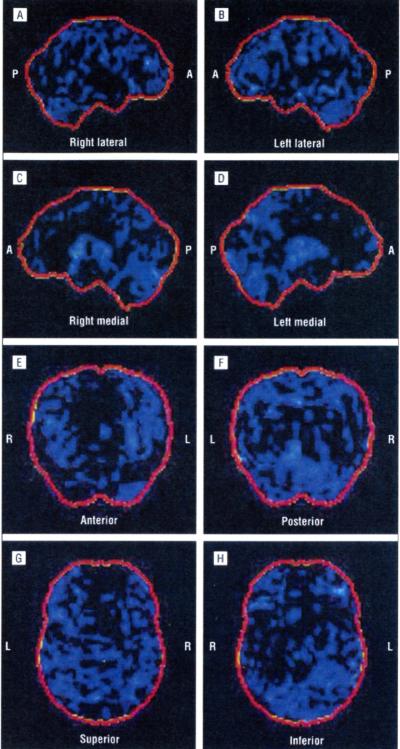
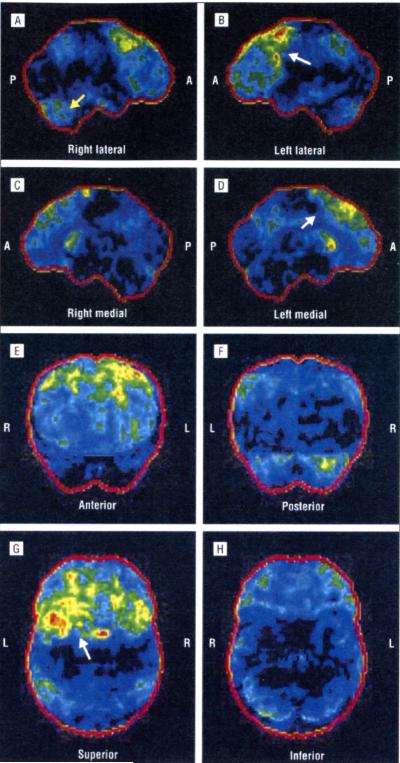
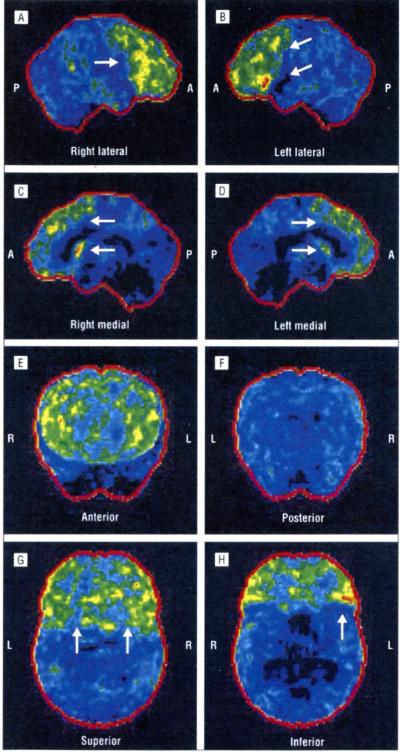
The p.Ala382Thr missense mutation of the TARDBP gene was absent in the 280 controls. It was present in all affected members of the 3 families for whom DNA was available. All affected members of the 3 families developed FTLD after the onset of ALS, confirmed by neuropsychological testing and hypometabolism in frontal associative areas assessed with fludeoxyglucose F 18 positron emission tomography and computed tomography.
Three apparently unrelated families with familial ALS carrying the p.Ala382Thr TARDBP missense mutation developed FTLD. In these families, FTLD cosegregates with ALS. Patients with ALS carrying TARDBP mutations may develop FTLD.
由TARDBP基因编码的TAR DNA结合蛋白43已被确定为伴或不伴肌萎缩侧索硬化(ALS)的额颞叶痴呆(FTLD)以及散发性ALS的主要病理蛋白。随后,在2%至3%的ALS患者(包括家族性和散发性ALS)中检测到了TARDBP基因突变。然而,据我们所知,仅有1例关于2名携带TARDBP基因突变的FTLD患者后来发展为运动神经元病的描述。
描述3个患有家族性ALS且携带TARDBP基因突变的意大利家庭中的认知异常情况。
设计、场所和参与者:对36例家族性非超氧化物歧化酶1基因(SOD1)ALS患者和280名健康对照进行基因、神经心理学和神经影像学分析。主要观察指标我们确定了3例携带TARDBP基因p.Ala382Thr错义突变且具有FTLD临床、神经影像学和神经心理学特征的家族性ALS索引病例。
280名对照中未检测到TARDBP基因的p.Ala382Thr错义突变。在可获取DNA的3个家庭的所有患病成员中均存在该突变。3个家庭的所有患病成员在ALS发病后均出现了FTLD,这通过神经心理学测试以及用氟脱氧葡萄糖F 18正电子发射断层扫描和计算机断层扫描评估的额叶联合区代谢减低得以证实。
3个明显不相关的携带p.Ala382Thr TARDBP错义突变的家族性ALS家庭出现了FTLD。在这些家庭中,FTLD与ALS共分离。携带TARDBP突变的ALS患者可能会发展为FTLD。