Division of Medicine, University College, London, UK.
Lancet. 2010 Sep 4;376(9743):794-801. doi: 10.1016/S0140-6736(10)60670-8. Epub 2010 Aug 25.
Complement is a key component of the innate immune system, and variation in genes that regulate its activation is associated with renal and other disease. We aimed to establish the genetic basis for a familial disorder of complement regulation associated with persistent microscopic haematuria, recurrent macroscopic haematuria, glomerulonephritis, and progressive renal failure.
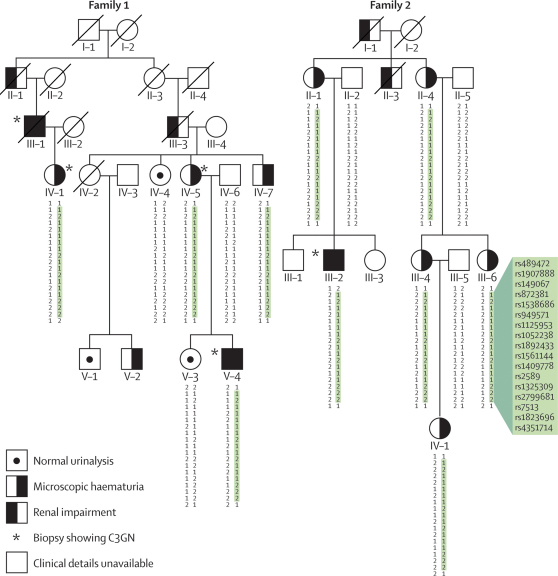
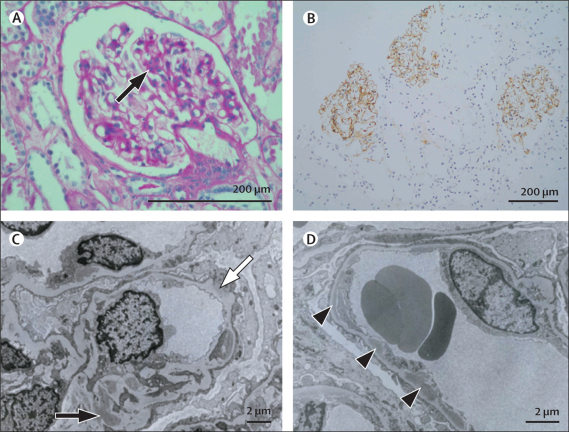
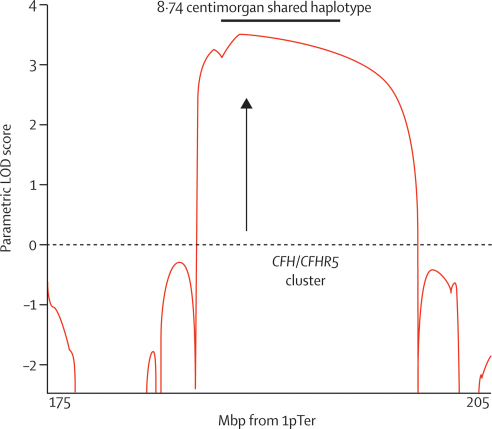
We sought patients from the West London Renal and Transplant Centre (London, UK) with unusual renal disease and affected family members as a method of identification of new genetic causes of kidney disease. Two families of Cypriot origin were identified in which renal disease was consistent with autosomal dominant transmission and renal biopsy of at least one individual showed C3 glomerulonephritis. A mutation was identified via a genome-wide linkage study and candidate gene analysis. A PCR-based diagnostic test was then developed and used to screen for the mutation in population-based samples and in individuals and families with renal disease.
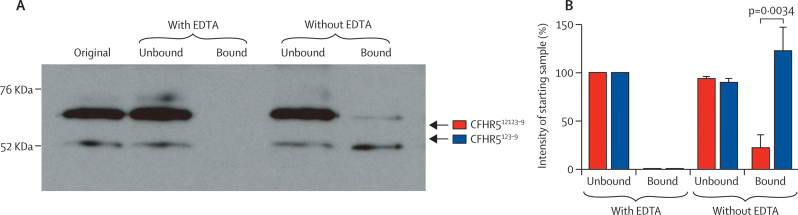
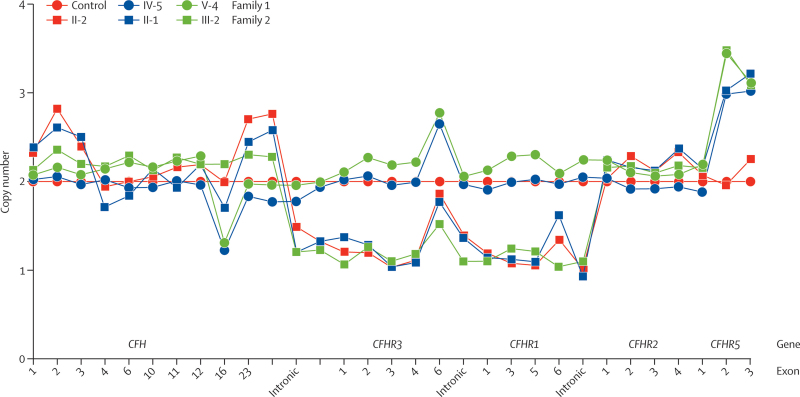
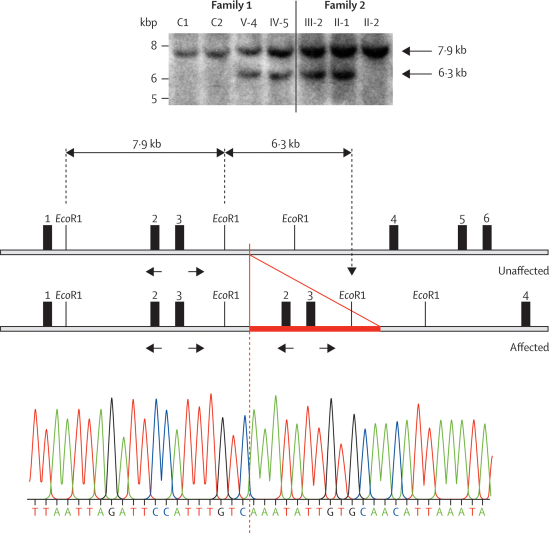
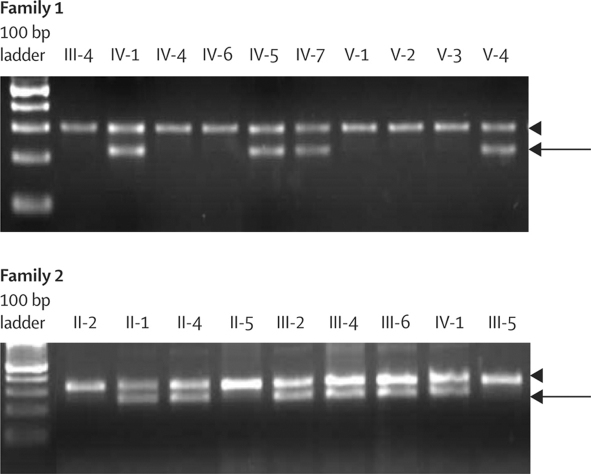
Occurrence of familial renal disease cosegregated with the same mutation in the complement factor H-related protein 5 gene (CFHR5). In a cohort of 84 Cypriots with unexplained renal disease, four had mutation in CFHR5. Overall, we identified 26 individuals with the mutation and evidence of renal disease from 11 ostensibly unrelated kindreds, including the original two families. A mutant CFHR5 protein present in patient serum had reduced affinity for surface-bound complement. We term this renal disease CFHR5 nephropathy.
CFHR5 nephropathy accounts for a substantial burden of renal disease in patients of Cypriot origin and can be diagnosed with a specific molecular test. The high risk of progressive renal disease in carriers of the CFHR5 mutation implies that isolated microscopic haematuria or recurrent macroscopic haematuria should not be regarded as a benign finding in individuals of Cypriot descent.
UK Medical Research Council and Wellcome Trust.
补体是先天免疫系统的关键组成部分,调节其激活的基因变异与肾脏和其他疾病有关。我们旨在确定与持续性镜下血尿、复发性肉眼血尿、肾小球肾炎和进行性肾功能衰竭相关的补体调节家族性疾病的遗传基础。
我们在英国伦敦西部肾脏和移植中心(London,UK)寻找患有不寻常肾脏疾病和受影响家族成员的患者,以此作为发现肾脏疾病新遗传病因的方法。确定了两个塞浦路斯血统的家族,其肾脏疾病符合常染色体显性遗传,至少有一名个体的肾脏活检显示 C3 肾小球肾炎。通过全基因组连锁研究和候选基因分析确定了一个突变。然后开发了一种基于 PCR 的诊断测试,并用于在基于人群的样本中以及在患有肾脏疾病的个体和家族中筛选该突变。
家族性肾脏疾病的发生与补体因子 H 相关蛋白 5 基因(CFHR5)中的相同突变密切相关。在一组 84 名原因不明的肾脏疾病的塞浦路斯人队列中,有 4 人携带 CFHR5 突变。总的来说,我们从 11 个看似无关的家族中发现了 26 名携带突变且有肾脏疾病证据的个体,包括最初的两个家族。患者血清中存在的突变 CFHR5 蛋白与表面结合的补体亲和力降低。我们将这种肾脏疾病称为 CFHR5 肾病。
CFHR5 肾病在塞浦路斯人起源的患者中占肾脏疾病的很大一部分,可以通过特定的分子测试进行诊断。CFHR5 突变携带者发生进行性肾脏疾病的风险很高,这意味着携带 CFHR5 突变的个体孤立性镜下血尿或复发性肉眼血尿不应被视为良性发现。
英国医学研究理事会和 Wellcome Trust。