McArdle Laboratory for Cancer Research, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, Wisconsin, United States of America.
PLoS One. 2010 Aug 16;5(8):e12213. doi: 10.1371/journal.pone.0012213.
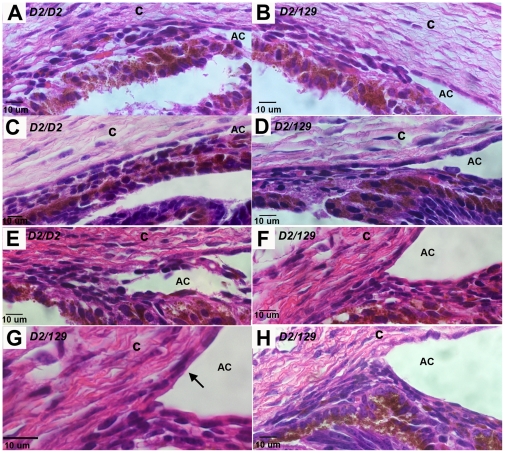
The PPCD1 mouse, a spontaneous mutant that arose in our mouse colony, is characterized by an enlarged anterior chamber resulting from metaplasia of the corneal endothelium and blockage of the iridocorneal angle by epithelialized corneal endothelial cells. The presence of stratified multilayered corneal endothelial cells with abnormal patterns of cytokeratin expression are remarkably similar to those observed in human posterior polymorphous corneal dystrophy (PPCD) and the sporadic condition, iridocorneal endothelial syndrome. Affected eyes exhibit epithelialized corneal endothelial cells, with inappropriate cytokeratin expression and proliferation over the iridocorneal angle and posterior cornea. We have termed this the "mouse PPCD1" phenotype and mapped the mouse locus for this phenotype, designated "Ppcd1", to a 6.1 Mbp interval on Chromosome 2, which is syntenic to the human Chromosome 20 PPCD1 interval. Inheritance of the mouse PPCD1 phenotype is autosomal dominant, with complete penetrance on the sensitive DBA/2J background and decreased penetrance on the C57BL/6J background. Comparative genome hybridization has identified a hemizygous 78 Kbp duplication in the mapped interval. The endpoints of the duplication are located in positions that disrupt the genes Csrp2bp and 6330439K17Rik and lead to duplication of the pseudogene LOC100043552. Quantitative reverse transcriptase-PCR indicates that expression levels of Csrp2bp and 6330439K17Rik are decreased in eyes of PPCD1 mice. Based on the observations of decreased gene expression levels, association with ZEB1-related pathways, and the report of corneal opacities in Csrp2bp(tm1a(KOMP)Wtsi) heterozygotes and embryonic lethality in nulls, we postulate that duplication of the 78 Kbp segment leading to haploinsufficiency of Csrp2bp is responsible for the mouse PPCD1 phenotype. Similarly, CSRP2BP haploinsufficiency may lead to human PPCD.
PPCD1 小鼠是我们的小鼠品系中自发出现的突变体,其特征是前房扩大,这是由于角膜内皮的化生和上皮化的角膜内皮细胞阻塞虹膜角膜角所致。具有异常细胞角蛋白表达模式的分层多层层状角膜内皮细胞与在人类后多形性角膜营养不良(PPCD)和散发性疾病、虹膜角膜内皮综合征中观察到的细胞非常相似。受影响的眼睛表现出上皮化的角膜内皮细胞,在虹膜角膜角和后角膜上表现出异常的细胞角蛋白表达和增殖。我们将这种表型称为“小鼠 PPCD1”表型,并将该表型的小鼠基因座命名为“Ppcd1”,定位于第 2 号染色体上的 6.1 Mbp 区间,与人类第 20 号染色体 PPCD1 区间是同源的。小鼠 PPCD1 表型的遗传是常染色体显性遗传,在敏感的 DBA/2J 背景下具有完全外显率,在 C57BL/6J 背景下外显率降低。比较基因组杂交鉴定出映射区间内的一个半合性 78 Kbp 重复。重复的端点位于破坏基因 Csrp2bp 和 6330439K17Rik 的位置,并导致假基因 LOC100043552 的重复。定量逆转录聚合酶链反应表明,PPCD1 小鼠眼睛中 Csrp2bp 和 6330439K17Rik 的表达水平降低。基于基因表达水平降低、与 ZEB1 相关途径的关联以及 Csrp2bp(tm1a(KOMP)Wtsi)杂合子中的角膜混浊报告和 nulls 中的胚胎致死性,我们推测导致 Csrp2bp 单倍不足的 78 Kbp 片段重复是导致小鼠 PPCD1 表型的原因。同样,CSRP2BP 单倍不足可能导致人类 PPCD。