Center for Neurodegeneration and Experimental Therapeutics, Department of Neurology, The University of Alabama at Birmingham, USA.
Mol Neurodegener. 2010 Oct 26;5:42. doi: 10.1186/1750-1326-5-42.
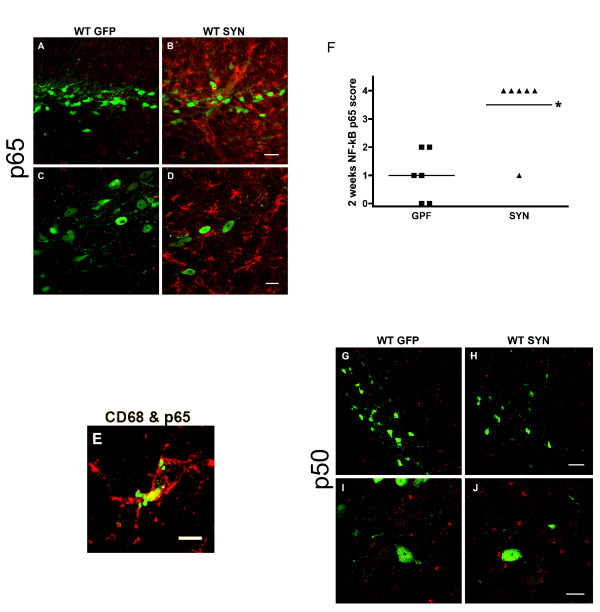
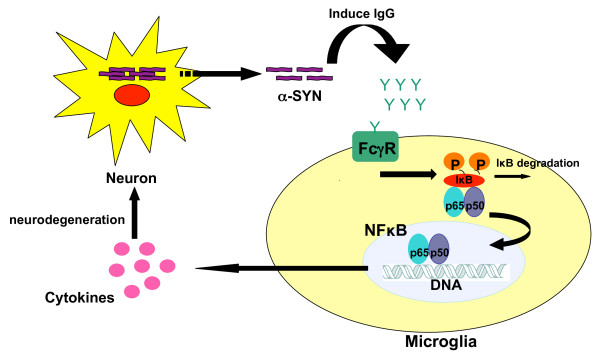
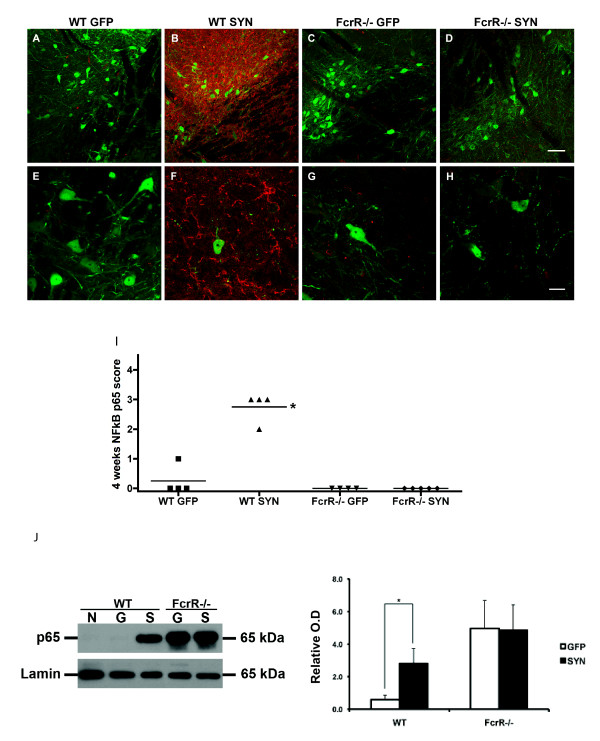
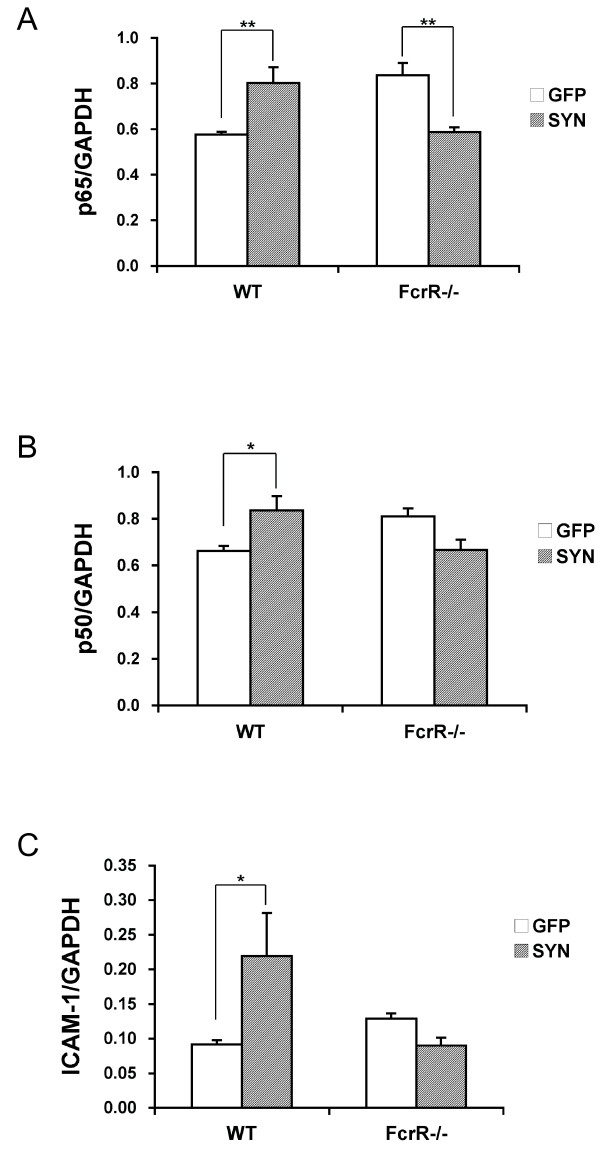
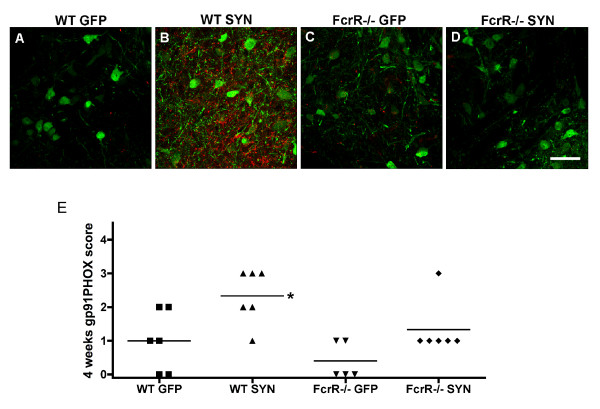
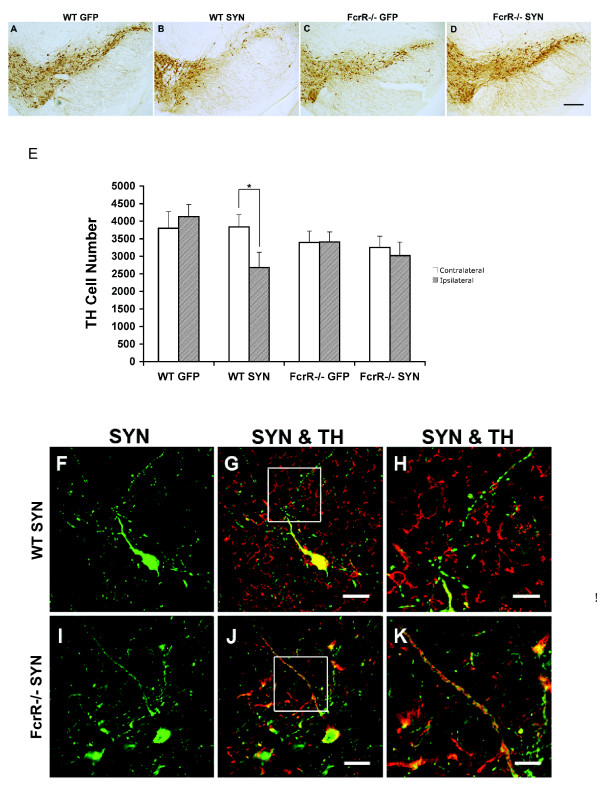
Overexpression of alpha-synuclein (α-SYN), a protein which plays an important role in the pathogenesis of Parkinson's disease (PD), triggers microglial activation and adaptive immune responses, and leads to neurodegeneration of dopaminergic (DA) neurons. We hypothesized a link between the humoral adaptive immune response and microglial activation in α-SYN induced neurodegeneration. To test this hypothesis, we employed adeno-associated virus serotype 2 (AAV2) to selectively over-express human α-SYN in the substantia nigra (SN) of wild-type mice and FcγR-/- mice, which lack high-affinity receptors for IgG. We found that in wild-type mice, α-SYN induced the expression of NF-κB p65 and pro-inflammatory molecules. In FcγR-/- mice, NF-κB activation was blocked and pro-inflammatory signaling was reduced. Microglial activation was examined using immunohistochemistry for gp91PHOX. At four weeks, microglia were strongly activated in wild-type mice, while microglial activation was attenuated in FcγR-/- mice. Dopaminergic neurodegeneration was examined using immunohistochemistry for tyrosine hydroxylase (TH) and unbiased stereology. α-SYN overexpression led to the appearance of dysmorphic neurites, and a loss of DA neurons in the SN in wild-type animals, while FcγR-/- mice did not exhibit neuritic change and were protected from α-SYN-induced neurodegeneration 24 weeks after injection. Our results suggest that the humoral adaptive immune response triggered by excess α-SYN plays a causative role in microglial activation through IgG-FcγR interaction. This involves NF-κB signaling, and leads to DA neurodegeneration. Therefore, blocking either FcγR signaling or specific intracellular signal transduction events downstream of FcγR-IgG interaction, such as NF-κB activation, may be viable therapeutic strategies in PD.
α-突触核蛋白(α-SYN)的过度表达在帕金森病(PD)的发病机制中起着重要作用,它触发小胶质细胞激活和适应性免疫反应,导致多巴胺能(DA)神经元的神经退行性变。我们假设体液适应性免疫反应与α-SYN 诱导的神经退行性变中小胶质细胞的激活之间存在联系。为了验证这一假设,我们使用腺相关病毒血清型 2(AAV2)选择性地在野生型小鼠和缺乏 IgG 高亲和力受体的 FcγR-/- 小鼠的黑质(SN)中过表达人 α-SYN。我们发现,在野生型小鼠中,α-SYN 诱导了 NF-κB p65 和促炎分子的表达。在 FcγR-/- 小鼠中,NF-κB 激活被阻断,促炎信号转导减少。使用针对 gp91PHOX 的免疫组织化学检查小胶质细胞的激活。在 4 周时,野生型小鼠中的小胶质细胞强烈激活,而 FcγR-/- 小鼠中的小胶质细胞激活减弱。使用针对酪氨酸羟化酶(TH)和无偏立体学的免疫组织化学检查 DA 神经元的退行性变。α-SYN 的过表达导致了形态异常的神经突的出现,并且在野生型动物的 SN 中 DA 神经元丢失,而 FcγR-/- 小鼠没有表现出神经突改变并且在注射后 24 周免受 α-SYN 诱导的神经退行性变的影响。我们的结果表明,由过量 α-SYN 触发的体液适应性免疫反应通过 IgG-FcγR 相互作用在小胶质细胞激活中起因果作用。这涉及 NF-κB 信号转导,并导致 DA 神经元退行性变。因此,阻断 FcγR 信号或 FcγR-IgG 相互作用下游的特定细胞内信号转导事件,例如 NF-κB 激活,可能是 PD 的可行治疗策略。