Department of Ophthalmology, Emory University School of Medicine, Atlanta, Georgia, USA.
Ophthalmology. 2011 Mar;118(3):558-63. doi: 10.1016/j.ophtha.2010.07.029. Epub 2010 Oct 30.
Autosomal-dominant optic atrophy (DOA) is one of the most common inherited optic neuropathies, and it is genetically heterogeneous, with mutations in both OPA1 and OPA3 known to cause disease. Approximately 60% of cases harbor OPA1 mutations, whereas OPA3 mutations have been reported in only 2 pedigrees with DOA and premature cataracts. The aim of this study was to determine the yield of OPA1 and OPA3 screening in a cohort of presumed DOA cases referred to a tertiary diagnostic laboratory.
Retrospective case series.
One hundred eighty-eight probands with bilateral optic atrophy referred for molecular genetic investigations at a tertiary diagnostic facility: 38 patients with an autosomal-dominant pattern of inheritance and 150 sporadic cases.
OPA1 and OPA3 genetic testing was initially performed using polymerase chain reaction-based sequencing methods. The presence of large-scale OPA1 and OPA3 genomic rearrangements was assessed further with a targeted comparative genomic hybridization microarray platform. The 3 primary Leber hereditary optic neuropathy (LHON) mutations, m.3460G→>A, m.11778G→A, and m.14484T→C, also were screened in all patients.
The proportion of patients with OPA1 and OPA3 pathogenic mutations. The clinical profile observed in molecularly confirmed DOA cases.
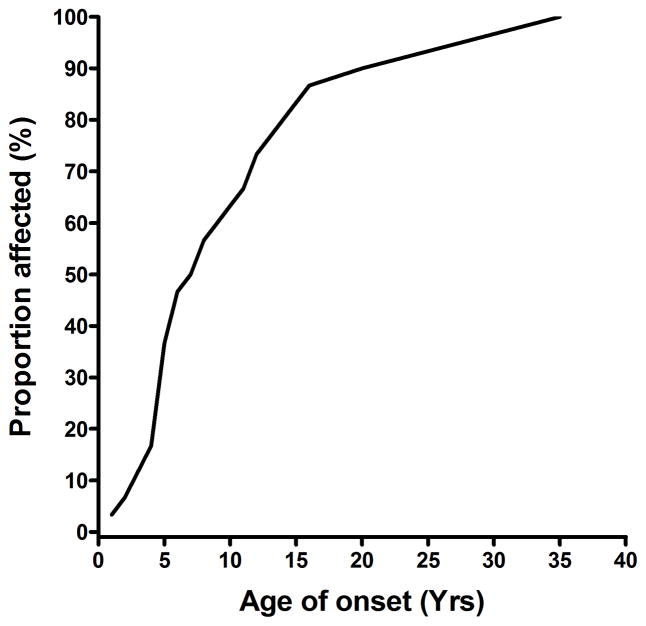
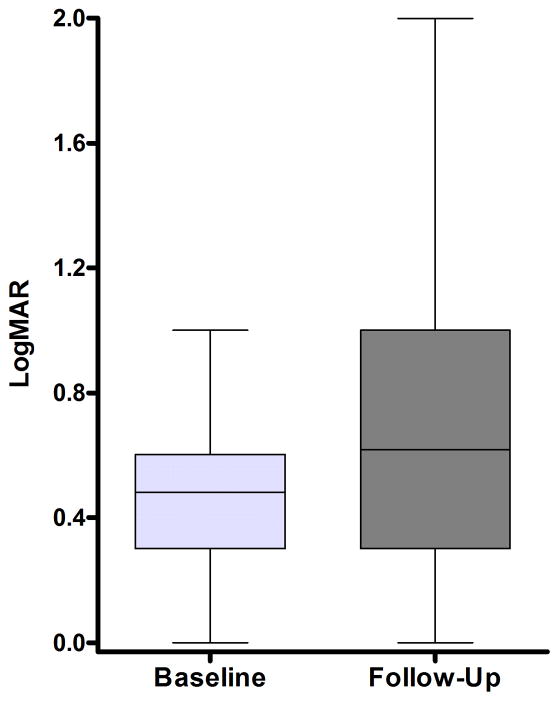
Twenty-one different OPA1 mutations were found in 27 (14.4%) of the 188 probands screened. The mutations included 6 novel pathogenic variants and the first reported OPA1 initiation codon mutation at c.1A→T. An OPA1 missense mutation, c.239A→G (p.Y80C), was identified in an 11-year-old black girl with optic atrophy and peripheral sensorimotor neuropathy in her lower limbs. The OPA1 detection rate was significantly higher among individuals with a positive family history of visual failure (50.0%) compared with sporadic cases (5.3%). The primary LHON screen was negative in the patient cohort, and additional molecular investigations did not reveal any large-scale OPA1 rearrangements or OPA3 genetic defects. The mean baseline visual acuity for the OPA1-positive group was 0.48 logarithm of the minimum angle of resolution (units mean Snellen equivalent, 20/61; range, 20/20-20/400; 95% confidence interval, 20/52-20/71), and visual deterioration occurred in 54.2% of patients during follow-up.
OPA1 mutations are the most common genetic defects identified in patients with suspected DOA, whereas OPA3 mutations are very rare in isolated optic atrophy cases.
常染色体显性视神经萎缩(DOA)是最常见的遗传性视神经病变之一,其遗传异质性,OPA1 和 OPA3 的突变已知可导致疾病。大约 60%的病例存在 OPA1 突变,而仅在 2 个具有 DOA 和早发性白内障的家系中报道了 OPA3 突变。本研究旨在确定在三级诊断实验室中对推定的 DOA 病例进行 OPA1 和 OPA3 筛查的结果。
回顾性病例系列。
188 名双侧视神经萎缩患者,在三级诊断机构进行分子遗传研究:38 名具有常染色体显性遗传模式的患者和 150 例散发性病例。
OPA1 和 OPA3 基因检测最初使用聚合酶链反应(PCR)测序方法进行。进一步使用靶向比较基因组杂交微阵列平台评估大的 OPA1 和 OPA3 基因组重排的存在。所有患者均筛查了 3 种主要的 Leber 遗传性视神经病变(LHON)突变,m.3460G→>A、m.11778G→A 和 m.14484T→C。
OPA1 和 OPA3 致病性突变患者的比例。经分子证实的 DOA 病例的临床特征。
在所筛选的 188 名患者中,发现了 27 名(14.4%)患者的 21 种不同的 OPA1 突变。这些突变包括 6 种新的致病性变体和首次报道的 c.1A→T 的 OPA1 起始密码子突变。在一名 11 岁的黑人女孩中发现了 OPA1 错义突变 c.239A→G(p.Y80C),该女孩患有视神经萎缩和下肢周围感觉运动神经病。OPA1 检测率在有阳性家族视觉衰竭史的个体中明显更高(50.0%),而在散发性病例中为 5.3%。患者队列的原发性 LHON 筛查呈阴性,进一步的分子研究未发现任何大的 OPA1 重排或 OPA3 遗传缺陷。OPA1 阳性组的基线平均视力为 0.48 对数最小角分辨率(单位为最小分辨角对数的倒数,Snellen 等价物 20/61;范围 20/20-20/400;95%置信区间 20/52-20/71),随访期间 54.2%的患者视力恶化。
OPA1 突变是疑似 DOA 患者中最常见的遗传缺陷,而 OPA3 突变在孤立性视神经萎缩病例中非常罕见。