Mitochondrial Research Group, The Medical School, Newcastle University, Newcastle upon Tyne, NE2 4HH, UK.
Brain. 2010 Mar;133(Pt 3):771-86. doi: 10.1093/brain/awq007. Epub 2010 Feb 15.
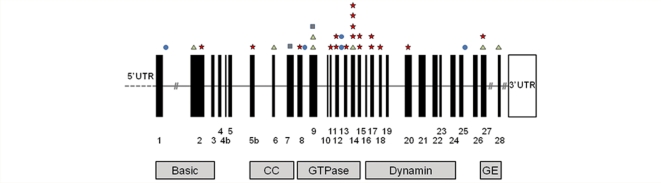
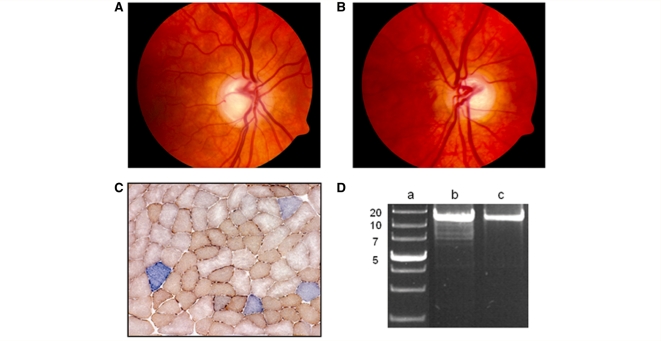
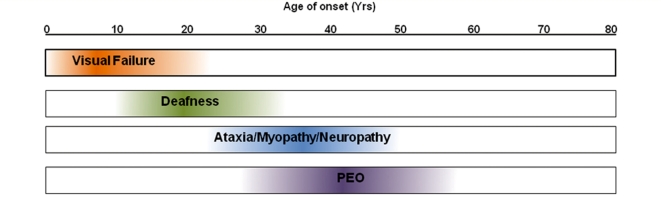
Additional neurological features have recently been described in seven families transmitting pathogenic mutations in OPA1, the most common cause of autosomal dominant optic atrophy. However, the frequency of these syndromal 'dominant optic atrophy plus' variants and the extent of neurological involvement have not been established. In this large multi-centre study of 104 patients from 45 independent families, including 60 new cases, we show that extra-ocular neurological complications are common in OPA1 disease, and affect up to 20% of all mutational carriers. Bilateral sensorineural deafness beginning in late childhood and early adulthood was a prominent manifestation, followed by a combination of ataxia, myopathy, peripheral neuropathy and progressive external ophthalmoplegia from the third decade of life onwards. We also identified novel clinical presentations with spastic paraparesis mimicking hereditary spastic paraplegia, and a multiple sclerosis-like illness. In contrast to initial reports, multi-system neurological disease was associated with all mutational subtypes, although there was an increased risk with missense mutations [odds ratio = 3.06, 95% confidence interval = 1.44-6.49; P = 0.0027], and mutations located within the guanosine triphosphate-ase region (odds ratio = 2.29, 95% confidence interval = 1.08-4.82; P = 0.0271). Histochemical and molecular characterization of skeletal muscle biopsies revealed the presence of cytochrome c oxidase-deficient fibres and multiple mitochondrial DNA deletions in the majority of patients harbouring OPA1 mutations, even in those with isolated optic nerve involvement. However, the cytochrome c oxidase-deficient load was over four times higher in the dominant optic atrophy + group compared to the pure optic neuropathy group, implicating a causal role for these secondary mitochondrial DNA defects in disease pathophysiology. Individuals with dominant optic atrophy plus phenotypes also had significantly worse visual outcomes, and careful surveillance is therefore mandatory to optimize the detection and management of neurological disability in a group of patients who already have significant visual impairment.
最近在七个传递 OPA1 致病性突变的家族中描述了其他神经学特征,OPA1 是常染色体显性视神经萎缩的最常见原因。然而,这些综合征“显性视神经萎缩加”变体的频率和神经受累的程度尚未确定。在这项针对来自 45 个独立家族的 104 名患者(包括 60 例新病例)的大型多中心研究中,我们表明,OPA1 疾病中外眼神经并发症很常见,影响到所有突变携带者的 20%。始于童年后期和成年早期的双侧感觉神经性耳聋是突出的表现,随后从第三十年开始出现共济失调、肌病、周围神经病和进行性眼外肌麻痹的组合。我们还发现了具有痉挛性截瘫样表现的新的临床表现,类似于遗传性痉挛性截瘫,以及多发性硬化样疾病。与最初的报告相反,多系统神经疾病与所有突变亚型有关,尽管错义突变的风险增加[优势比=3.06,95%置信区间=1.44-6.49;P=0.0027],并且突变位于鸟嘌呤三磷酸酶区域内(优势比=2.29,95%置信区间=1.08-4.82;P=0.0271)。骨骼肌活检的组织化学和分子特征表明,大多数携带 OPA1 突变的患者存在细胞色素 c 氧化酶缺陷纤维和多种线粒体 DNA 缺失,即使是孤立的视神经受累患者也是如此。然而,在显性视神经萎缩+组中,细胞色素 c 氧化酶缺陷负荷比单纯视神经病变组高四倍以上,这表明这些继发性线粒体 DNA 缺陷在疾病病理生理学中起因果作用。具有显性视神经萎缩加表型的个体的视觉预后也明显较差,因此必须进行仔细监测,以优化一组已经存在明显视力障碍的患者的神经功能障碍的检测和管理。