University of Pittsburgh, Department of Developmental Biology, 8111 Rangos Research Center, 530 45th Street, Pittsburgh, PA 15201, USA.
Dis Model Mech. 2011 Jan;4(1):43-56. doi: 10.1242/dmm.006262. Epub 2010 Nov 2.
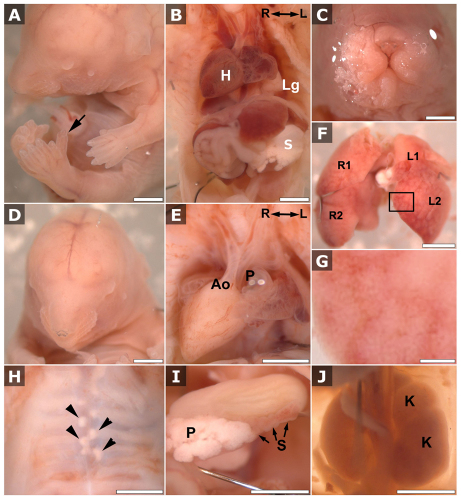
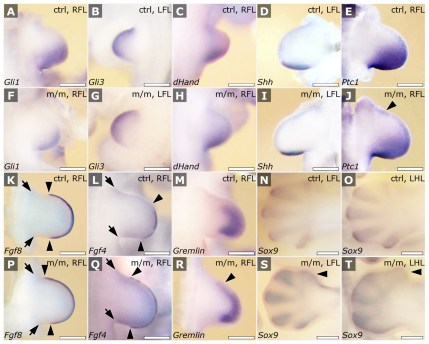
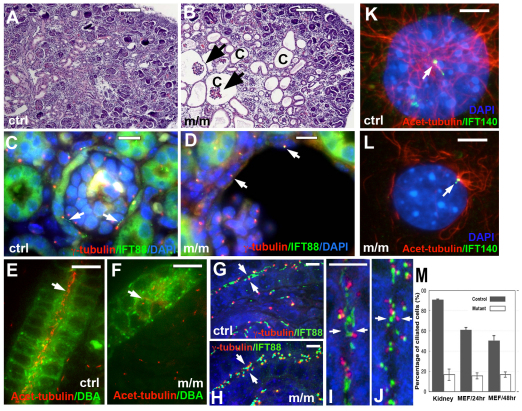
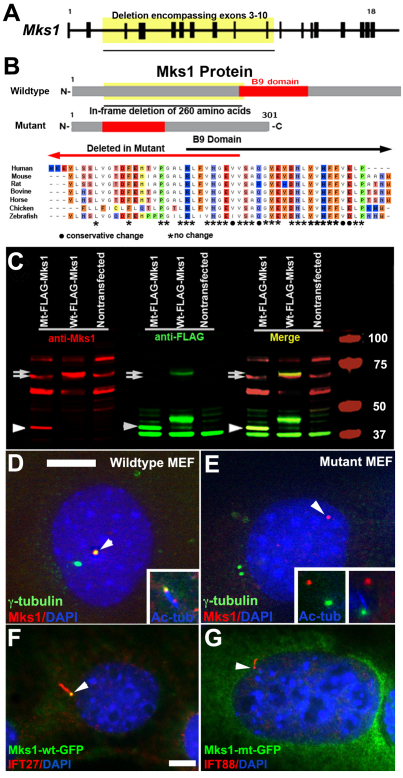
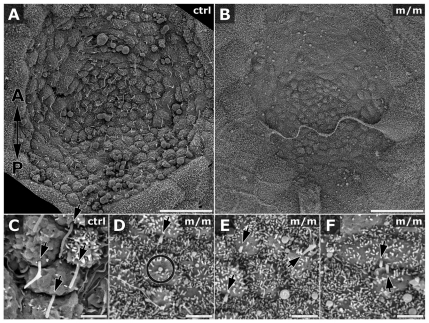
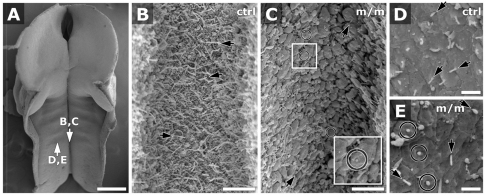
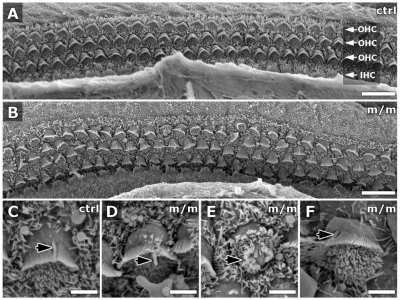
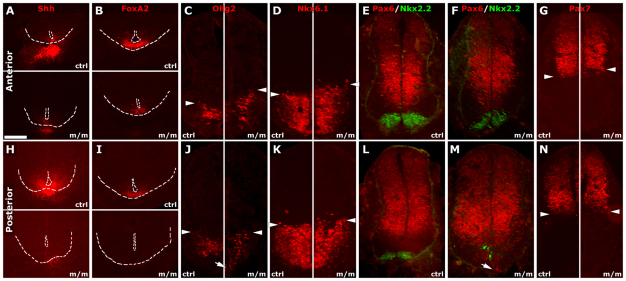
Meckel-Gruber syndrome (MKS) is a recessive disorder resulting in multiple birth defects that are associated with mutations affecting ciliogenesis. We recovered a mouse mutant with a mutation in the Mks1 gene (Mks1(del64-323)) that caused a 260-amino-acid deletion spanning nine amino acids in the B9 domain, a protein motif with unknown function conserved in two other basal body proteins. We showed that, in wild-type cells, Mks1 was localized to the mother centriole from which the cilium was generated. However, in mutant Mks1(del64-323) cells, Mks1 was not localized to the centriole, even though it maintained a punctate distribution. Resembling MKS patients, Mks1 mutants had craniofacial defects, polydactyly, congenital heart defects, polycystic kidneys and randomized left-right patterning. These defects reflected disturbance of functions subserved by motile and non-motile cilia. In the kidney, glomerular and tubule cysts were observed along with short cilia, and cilia were reduced in number to a near-complete loss. Underlying the left-right patterning defects were fewer and shorter nodal cilia, and analysis with fluorescent beads showed no directional flow at the embryonic node. In the cochlea, the stereocilia were mal-patterned, with the kinocilia being abnormally positioned. Together, these defects suggested disruption of planar cell polarity, which is known to regulate node, kidney and cochlea development. In addition, we also showed that Shh signaling was disrupted. Thus, in the neural tube, the floor plate was not specified posteriorly even as expression of the Shh mediator Gli2 increased. By contrast, the Shh signaling domain was expanded in the anterior neural tube and anterior limb bud, consistent with reduced Gli3-repressor (Gli3R) function. The latter probably accounted for the preaxial digit duplication exhibited by the Mks1(del64-323) mutants. Overall, these findings indicate that centriole localization of Mks1 is required for ciliogenesis of motile and non-motile cilia, but not for centriole assembly. On the basis of these results, we hypothesize a role for the B9 domain in mother centriole targeting, a possibility that warrants further future investigations.
梅克尔-格鲁伯综合征(MKS)是一种隐性疾病,导致多种出生缺陷,这些缺陷与影响纤毛发生的基因突变有关。我们从 Mks1 基因突变(Mks1(del64-323))的小鼠突变体中恢复了一个突变体,该突变体导致跨越 9 个氨基酸的 B9 结构域的 260 个氨基酸缺失,该结构域是一种具有未知功能的蛋白基序,在另外两种基底体蛋白中保守。我们表明,在野生型细胞中,Mks1 定位于生成纤毛的母中心粒。然而,在突变体 Mks1(del64-323)细胞中,Mks1 未定位于中心粒,尽管它保持点状分布。与 MKS 患者相似,Mks1 突变体具有颅面缺陷、多指、先天性心脏缺陷、多囊肾和随机的左右图案化。这些缺陷反映了运动和非运动纤毛的功能障碍。在肾脏中,观察到肾小球和肾小管囊肿以及短纤毛,纤毛数量减少到几乎完全丧失。左右图案化缺陷的基础是更少和更短的 nodal 纤毛,并且用荧光珠分析显示胚胎节点没有定向流动。在耳蜗中,静纤毛模式异常,动纤毛位置异常。总之,这些缺陷表明平面细胞极性的破坏,这已知调节节点、肾脏和耳蜗发育。此外,我们还表明 Shh 信号被破坏。因此,在神经管中,即使 Shh 介质 Gli2 的表达增加,后基板也不能被指定。相比之下,Shh 信号域在前神经管和前肢芽中扩展,与减少的 Gli3 抑制剂(Gli3R)功能一致。后者可能是 Mks1(del64-323)突变体表现出的前轴指重复的原因。总的来说,这些发现表明 Mks1 的中心粒定位对于运动和非运动纤毛的发生是必需的,但对于中心粒组装不是必需的。基于这些结果,我们假设 B9 结构域在母中心粒靶向中起作用,这是一个值得进一步未来研究的可能性。