Center for Bioinformatics Tübingen (ZBIT), University of Tübingen, Tübingen, Germany.
PLoS One. 2010 Nov 30;5(11):e13876. doi: 10.1371/journal.pone.0013876.
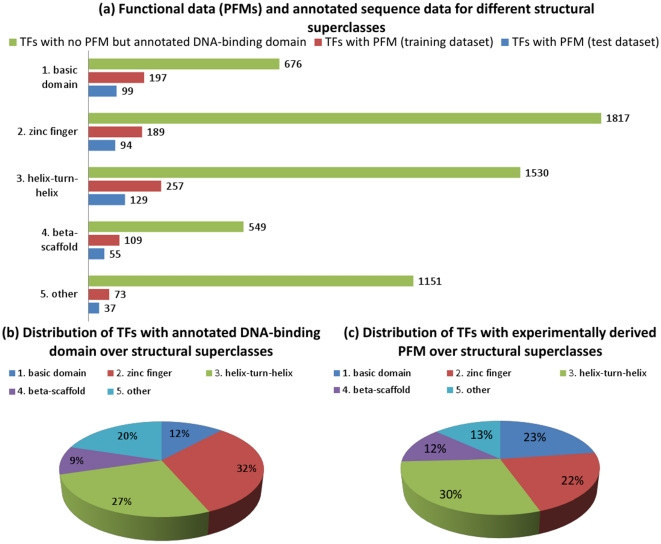
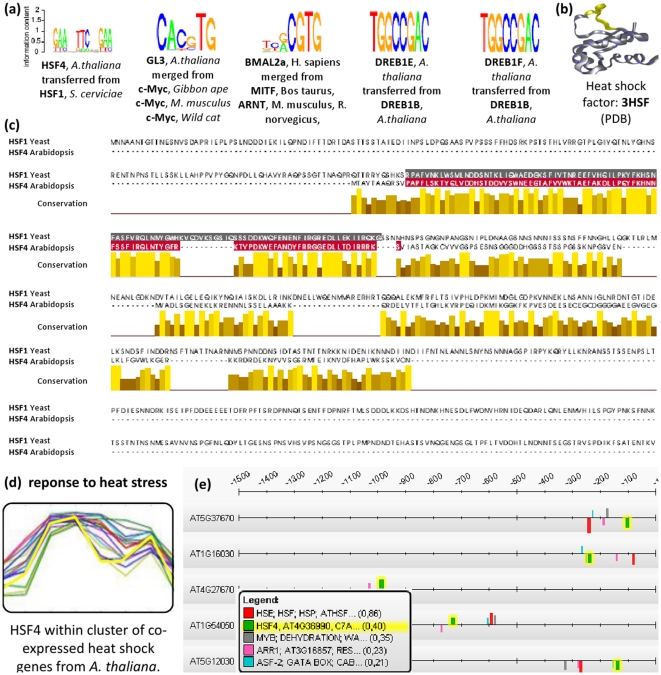
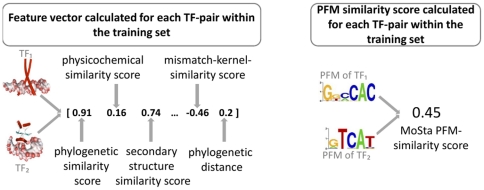
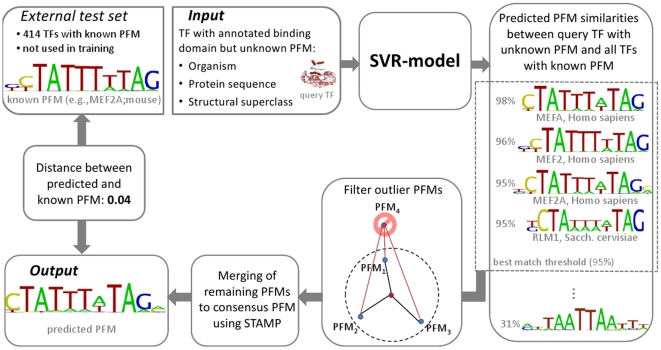
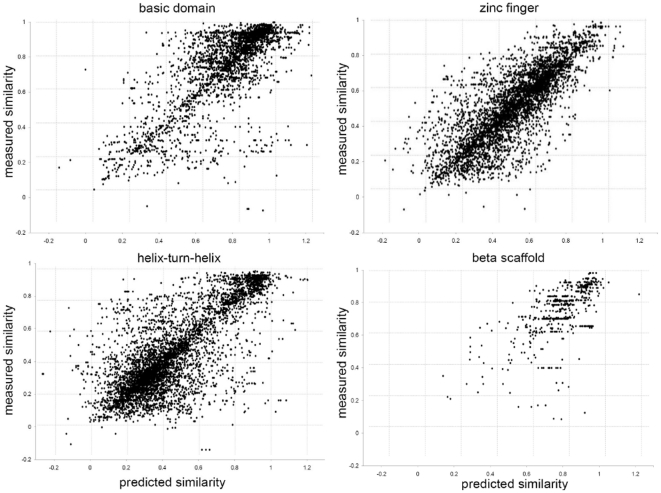
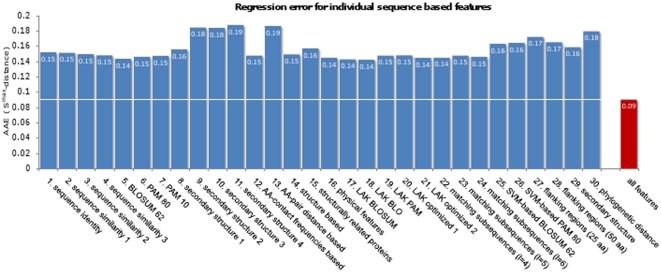
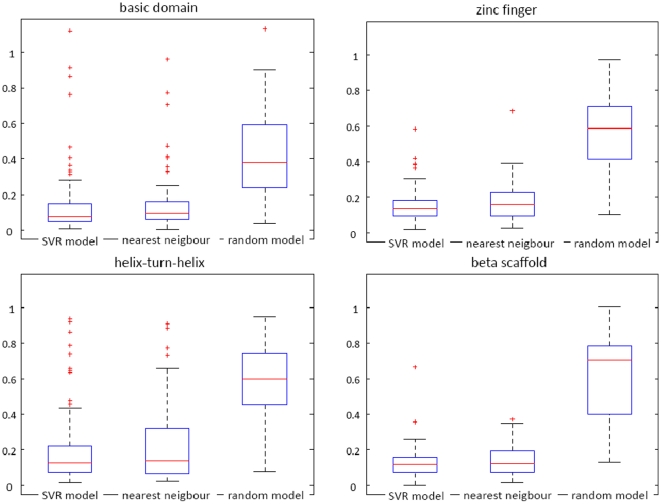
Today, annotated amino acid sequences of more and more transcription factors (TFs) are readily available. Quantitative information about their DNA-binding specificities, however, are hard to obtain. Position frequency matrices (PFMs), the most widely used models to represent binding specificities, are experimentally characterized only for a small fraction of all TFs. Even for some of the most intensively studied eukaryotic organisms (i.e., human, rat and mouse), roughly one-sixth of all proteins with annotated DNA-binding domain have been characterized experimentally. Here, we present a new method based on support vector regression for predicting quantitative DNA-binding specificities of TFs in different eukaryotic species. This approach estimates a quantitative measure for the PFM similarity of two proteins, based on various features derived from their protein sequences. The method is trained and tested on a dataset containing 1 239 TFs with known DNA-binding specificity, and used to predict specific DNA target motifs for 645 TFs with high accuracy.
如今,越来越多的转录因子 (TF) 的注释氨基酸序列都可以轻松获得。然而,关于它们的 DNA 结合特异性的定量信息却很难获取。位置频率矩阵 (PFM) 是最广泛用于表示结合特异性的模型,但仅对一小部分 TF 进行了实验表征。即使对于一些研究最深入的真核生物(即人类、大鼠和小鼠),也只有大约六分之一的具有注释 DNA 结合域的蛋白质进行了实验表征。在这里,我们提出了一种新的基于支持向量回归的方法,用于预测不同真核生物中 TF 的定量 DNA 结合特异性。该方法基于从蛋白质序列中提取的各种特征,估计两个蛋白质的 PFM 相似性的定量度量。该方法在包含 1239 个具有已知 DNA 结合特异性的 TF 的数据集上进行训练和测试,并用于高精度地预测 645 个具有高特异性的 TF 的特定 DNA 靶标基序。