Department of Biology, University of Bergen, Post box 7800, N-5020 Bergen, Norway.
BMC Vet Res. 2011 Jan 24;7:5. doi: 10.1186/1746-6148-7-5.
Since Francisella noatunensis was first isolated from cultured Atlantic cod in 2004, it has emerged as a global fish pathogen causing disease in both warm and cold water species. Outbreaks of francisellosis occur in several important cultured fish species making a correct management of this disease a matter of major importance. Currently there are no vaccines or treatments available. A strain typing system for use in studies of F. noatunensis epizootics would be an important tool for disease management. However, the high genetic similarity within the Francisella spp. makes strain typing difficult, but such typing of the related human pathogen Francisella tullarensis has been performed successfully by targeting loci with higher genetic variation than the traditional signature sequences. These loci are known as Variable Numbers of Tandem Repeat (VNTR). The aim of this study is to identify possible useful VNTRs in the genome of F. noatunensis.
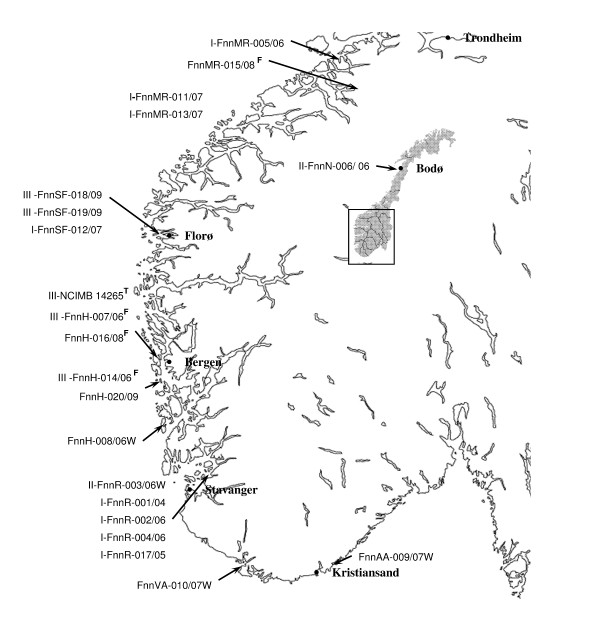
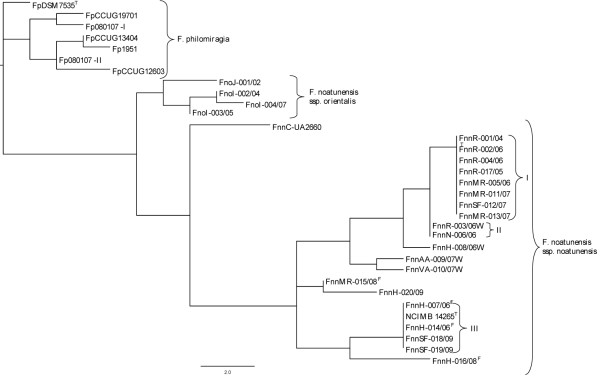
Seven polymorphic VNTR loci were identified in the preliminary genome sequence of F. noatunensis ssp. noatunensis GM2212 isolate. These VNTR-loci were sequenced in F. noatunensis isolates collected from Atlantic cod (Gadus morhua) from Norway (n = 21), Three-line grunt (Parapristipoma trilineatum) from Japan (n = 1), Tilapia (Oreochromis spp.) from Indonesia (n = 3) and Atlantic salmon (Salmo salar) from Chile (n = 1). The Norwegian isolates presented in this study show both nine allelic profiles and clades, and that the majority of the farmed isolates belong in two clades only, while the allelic profiles from wild cod are unique.
VNTRs can be used to separate isolates belonging to both subspecies of F. noatunensis. Low allelic diversity in F. noatunensis isolates from outbreaks in cod culture compared to isolates wild cod, indicate that transmission of these isolates may be a result of human activity. The sequence based MLVA system presented in this study should provide a good starting point for further development of a genotyping system that can be used in studies of epizootics and disease management of francisellosis.
自 2004 年弗朗西斯氏菌首次从大西洋养殖鳕鱼中分离出来以来,它已成为一种全球性的鱼类病原体,可感染温水和冷水物种。几种重要养殖鱼类的弗朗西斯菌病暴发时有发生,因此正确管理这种疾病至关重要。目前尚无疫苗或治疗方法。一种用于弗朗西斯氏菌流行性病学研究的菌株分型系统将是疾病管理的重要工具。然而,弗朗西斯菌属内的遗传相似度很高,使得菌株分型变得困难,但对相关人类病原体弗朗西斯菌 tularensis 的这种分型已通过针对遗传变异高于传统特征序列的基因座成功完成。这些基因座被称为可变数量串联重复(VNTR)。本研究的目的是确定弗朗西斯氏菌 GM2212 分离株基因组中可能有用的 VNTR。
在弗朗西斯氏菌 ssp.noatunensis GM2212 分离株的初步基因组序列中鉴定出 7 个多态性 VNTR 基因座。在从挪威大西洋鳕鱼(Gadus morhua)(n = 21)、日本三线鲷(Parapristipoma trilineatum)(n = 1)、印度尼西亚罗非鱼(Oreochromis spp.)(n = 3)和智利大西洋鲑(Salmo salar)(n = 1)中收集的弗朗西斯氏菌分离株中对这些 VNTR 基因座进行了测序。本研究中呈现的挪威分离株显示出 9 种等位基因谱和 2 个聚类,并且大多数养殖分离株仅属于 2 个聚类,而野生鳕鱼的等位基因谱是独特的。
VNTR 可用于分离属于弗朗西斯氏菌两种亚种的分离株。与野生鳕鱼分离株相比,鳕鱼养殖暴发中弗朗西斯氏菌分离株的等位基因多样性较低,表明这些分离株的传播可能是人类活动的结果。本研究中提出的基于序列的 MLVA 系统应为进一步开发用于弗朗西斯氏菌病流行性病学和疾病管理研究的基因分型系统提供良好的起点。