Gadhe Changdev G, Madhavan Thirumurthy, Kothandan Gugan, Cho Seung Joo
Departments of Bio-New Drug Development, College of Medicine, Chosun University, 375 Seosuk-dong, Dong-gu, Gwangju 501-759, Korea.
BMC Struct Biol. 2011 Jan 26;11:5. doi: 10.1186/1472-6807-11-5.
Multidrug resistance (MDR) is a major obstacle in cancer chemotherapy. The drug efflux by a transport protein is the main reason for MDR. In humans, MDR mainly occurs when the ATP-binding cassette (ABC) family of proteins is overexpressed simultaneously. P-glycoprotein (P-gp) is most commonly associated with human MDR; it utilizes energy from adenosine triphosphate (ATP) to transport a number of substrates out of cells against concentration gradients. By the active transport of substrates against concentration gradients, intracellular concentrations of substrates are decreased. This leads to the cause of failure in cancer chemotherapy.
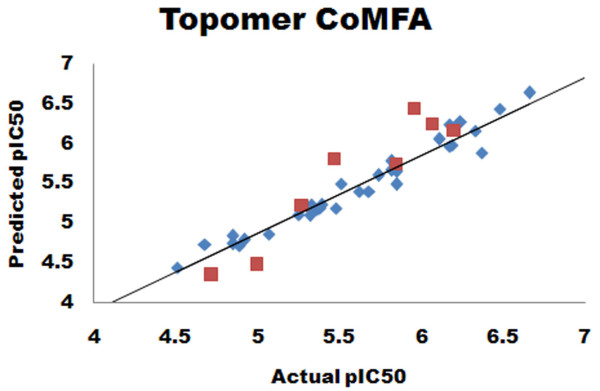
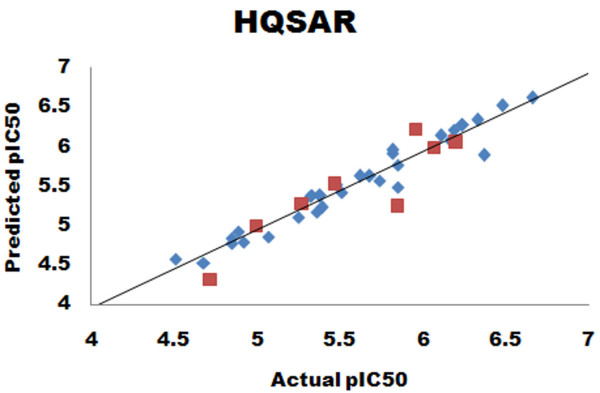
Herein, we report Topomer CoMFA (Comparative Molecular Field Analysis) and HQSAR (Hologram Quantitative Structure Activity Relationship) models for third generation MDR modulators. The Topomer CoMFA model showed good correlation between the actual and predicted values for training set molecules. The developed model showed cross validated correlation coefficient (q2) = 0.536 and non-cross validated correlation coefficient (r2) = 0.975 with eight components. The best HQSAR model (q2 = 0.777, r2 = 0.956) with 5-8 atom counts was used to predict the activity of test set compounds. Both models were validated using test set compounds, and gave a good predictive values of 0.604 and 0.730.
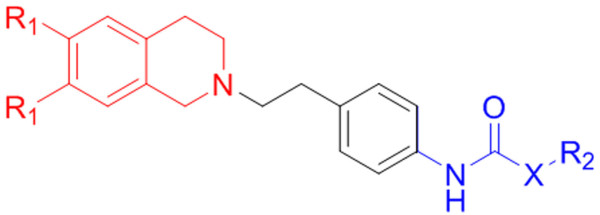
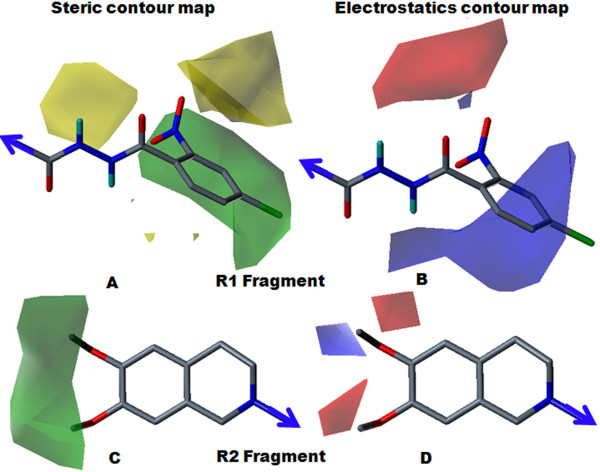
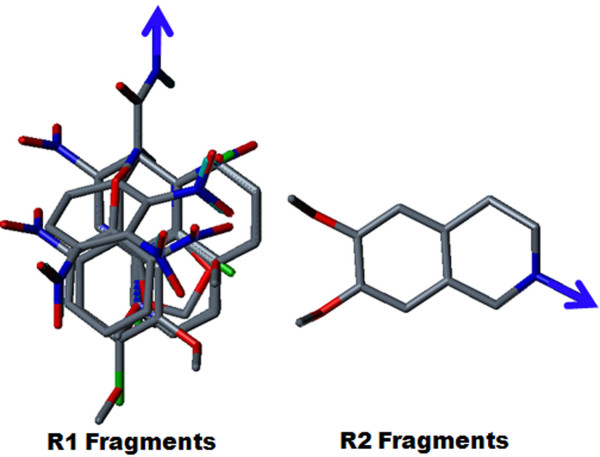
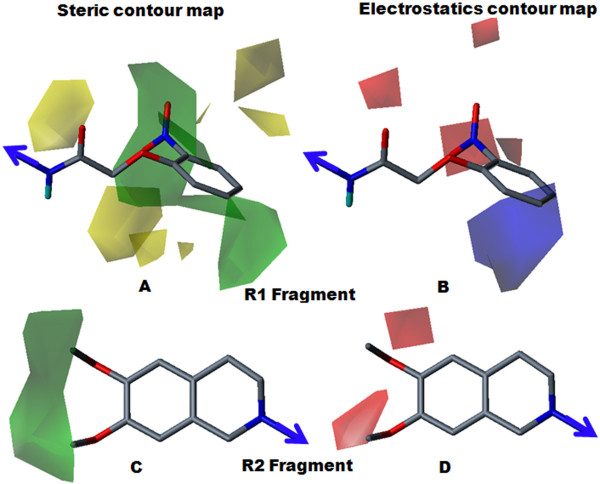
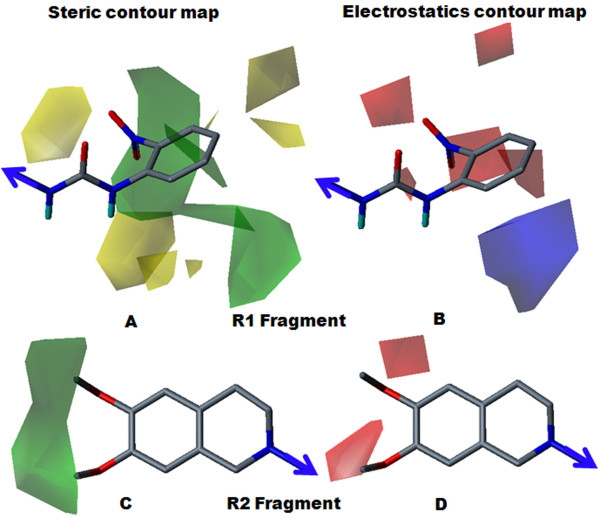
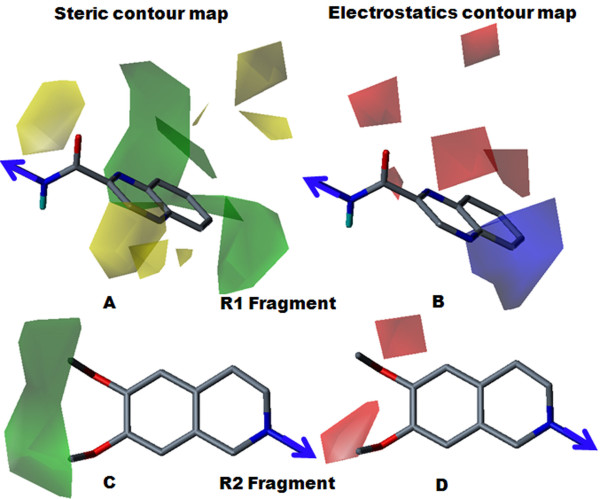
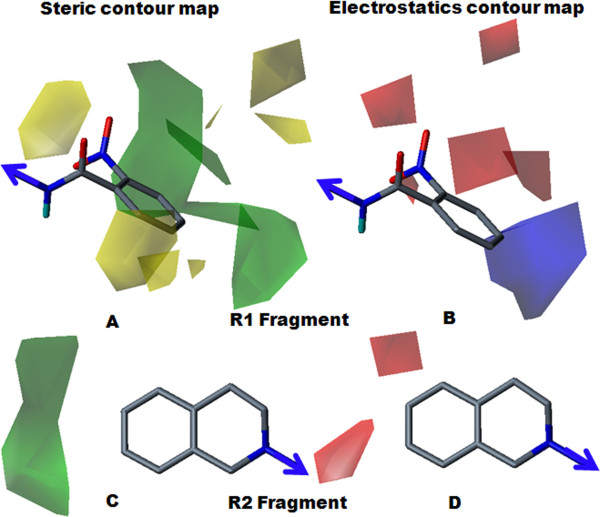
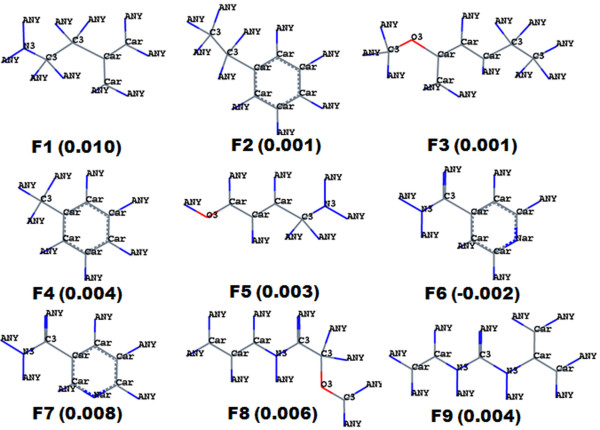
The contour map near R1 indicates that substitution of a bulkier and polar group to the ortho position of the benzene ring enhances the inhibitory effect. This explains why compounds with a nitro group have good inhibitory potency. Molecular fragment analyses shed light on some essential structural and topological features of third generation MDR modulators. Fragments analysis showed that the presence of tertiary nitrogen, a central phenyl ring and an aromatic dimethoxy group contributed to the inhibitory effect. Based on contour map information and fragment information, five new molecules with variable R1 substituents were designed. The activity of these designed molecules was predicted by the Topomer CoMFA and HQSAR models. The novel compounds showed higher potency than existing compounds.
多药耐药性(MDR)是癌症化疗中的主要障碍。转运蛋白介导的药物外排是多药耐药的主要原因。在人类中,多药耐药主要发生在ATP结合盒(ABC)蛋白家族同时过度表达时。P-糖蛋白(P-gp)与人类多药耐药最为相关;它利用三磷酸腺苷(ATP)提供的能量将多种底物逆浓度梯度转运出细胞。通过将底物逆浓度梯度主动转运,底物的细胞内浓度降低。这导致了癌症化疗失败的原因。
在此,我们报告了第三代多药耐药调节剂的Topomer CoMFA(比较分子场分析)和HQSAR(全息定量构效关系)模型。Topomer CoMFA模型显示训练集分子的实际值与预测值之间具有良好的相关性。所开发的模型显示交叉验证相关系数(q2)= 0.536,非交叉验证相关系数(r2)= 0.975,有八个成分。最佳的HQSAR模型(q2 = 0.777,r2 = 0.956),原子数为5 - 8,用于预测测试集化合物的活性。两个模型均使用测试集化合物进行验证,预测值分别为0.604和0.730,效果良好。
R1附近的等高线图表明,在苯环邻位取代一个更大的极性基团可增强抑制作用。这解释了为什么含硝基的化合物具有良好的抑制效力。分子片段分析揭示了第三代多药耐药调节剂的一些基本结构和拓扑特征。片段分析表明叔氮、中心苯环和芳族二甲氧基的存在有助于抑制作用。基于等高线图信息和片段信息,设计了五个具有可变R1取代基的新分子。这些设计分子的活性通过Topomer CoMFA和HQSAR模型进行预测。新化合物显示出比现有化合物更高的效力。