Department of Physiology and Tulane Renal Hypertension and Renal Center , Tulane University, School of Medicine, New Orleans, LA 70112, USa.
Hypertension. 2011 Mar;57(3):594-9. doi: 10.1161/HYPERTENSIONAHA.110.165902. Epub 2011 Jan 31.
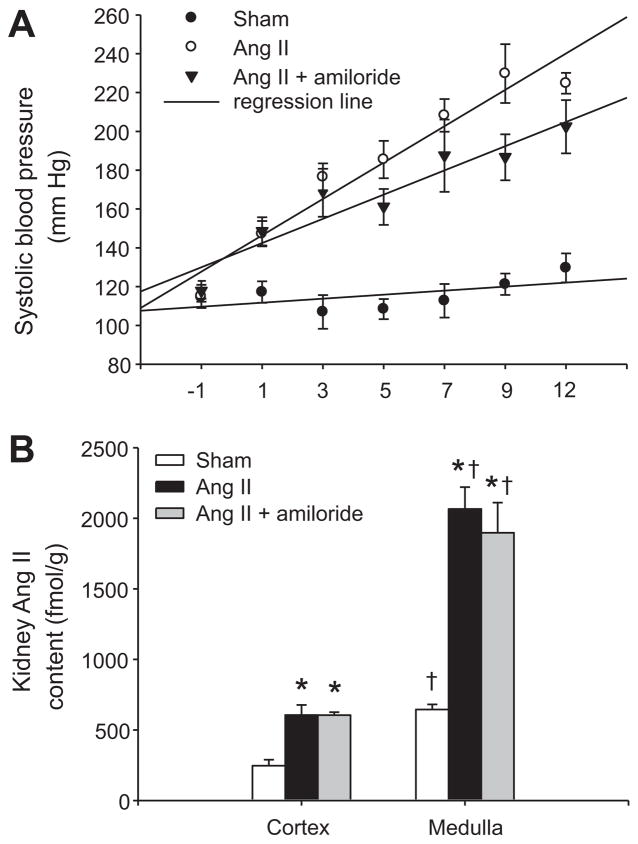
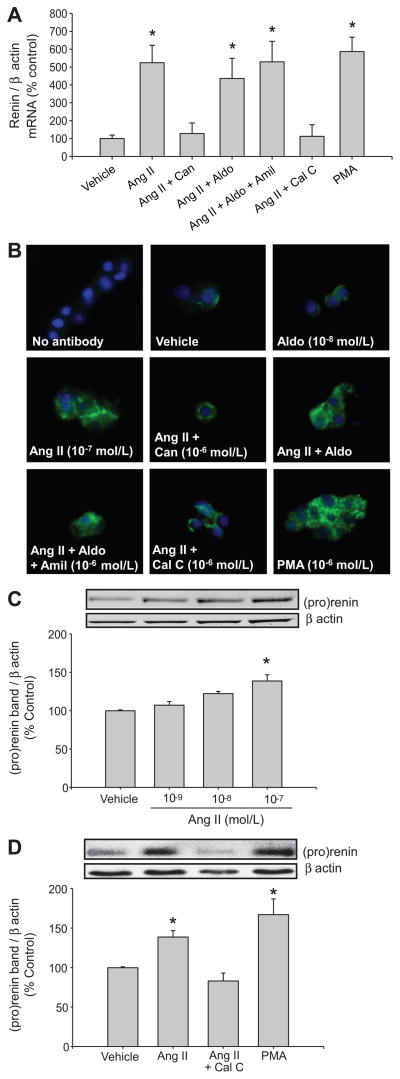
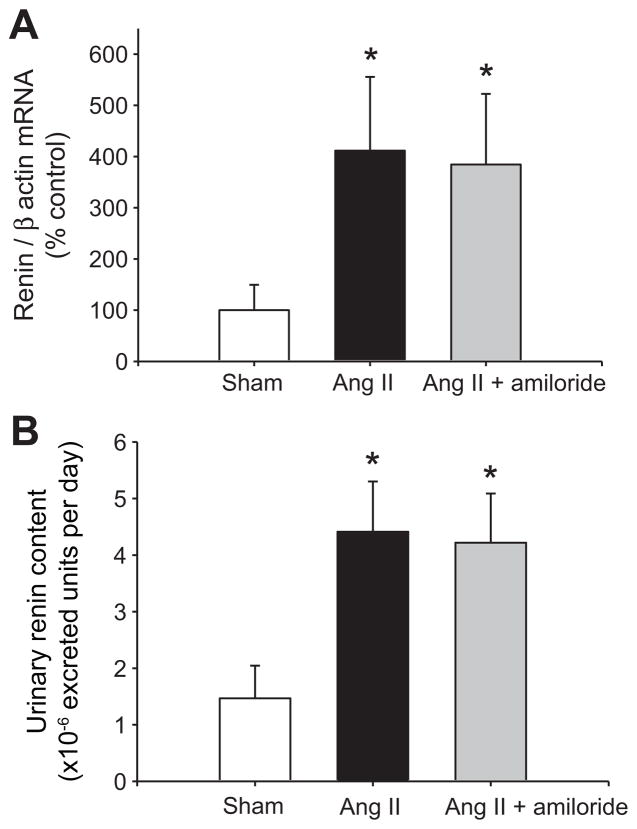
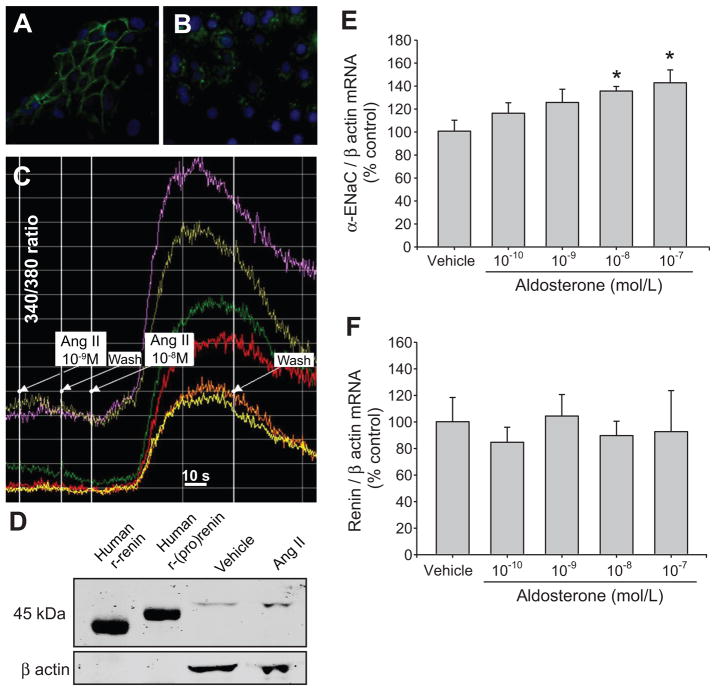
Collecting duct (CD) renin is stimulated by angiotensin (Ang) II, providing a pathway for Ang I generation and further conversion to Ang II. Ang II stimulates the epithelial sodium channel via the Ang II type 1 receptor and increases mineralocorticoid receptor activity attributed to increased aldosterone release. Our objective was to determine whether CD renin augmentation is mediated directly by Ang II type 1 receptor or via the epithelial sodium channel and mineralocorticoid receptor. In vivo studies examined the effects of epithelial sodium channel blockade (amiloride; 5 mg/kg per day) on CD renin expression and urinary renin content in Ang II-infused rats (80 ng/min, 2 weeks). Ang II infusion increased systolic blood pressure, medullary renin mRNA, urinary renin content, and intrarenal Ang II levels. Amiloride cotreatment did not alter these responses despite a reduction in the rate of progression of systolic blood pressure. In primary cultures of inner medullary CD cells, renin mRNA and (pro)renin protein levels increased with Ang II (100 nmol/L), and candesartan (Ang II type 1 receptor antagonist) prevented this effect. Aldosterone (10(-10) to 10(-7) mol/L) with or without amiloride did not modify the upregulation of renin mRNA in Ang II-treated cells. However, inhibition of protein kinase C with calphostin C prevented the Ang II-mediated increases in renin mRNA and (pro)renin protein levels. Furthermore, protein kinase C activation with phorbol 12-myristate 13-acetate increased renin expression to the same extent as Ang II. These data indicate that an Ang II type 1 receptor-mediated increase in CD renin is induced directly by Ang II via the protein kinase C pathway and that this regulation is independent of mineralocorticoid receptor activation or epithelial sodium channel activity.
收集管 (CD) 肾素受血管紧张素 (Ang) II 的刺激,为 Ang I 的生成和进一步转化为 Ang II 提供了途径。Ang II 通过 Ang II 型 1 受体刺激上皮钠通道,增加醛固酮释放导致的矿物皮质激素受体活性。我们的目的是确定 CD 肾素的增加是否是由 Ang II 型 1 受体直接介导,还是通过上皮钠通道和矿物皮质激素受体介导。体内研究检测了上皮钠通道阻断剂(阿米洛利;每天 5mg/kg)对 Ang II 输注大鼠(80ng/min,2 周)CD 肾素表达和尿肾素含量的影响。Ang II 输注增加了收缩压、髓质肾素 mRNA、尿肾素含量和肾内 Ang II 水平。尽管阿米洛利的联合治疗降低了收缩压的进展速度,但并没有改变这些反应。在内髓 CD 细胞的原代培养中,肾素 mRNA 和 (pro) 肾素蛋白水平随着 Ang II(100nmol/L)的增加而增加,坎地沙坦(Ang II 型 1 受体拮抗剂)阻止了这种作用。醛固酮(10(-10) 至 10(-7)mol/L)无论是否联合阿米洛利都不会改变 Ang II 处理细胞中肾素 mRNA 的上调。然而,钙调蛋白抑制剂 calphostin C 抑制蛋白激酶 C 可阻止 Ang II 介导的肾素 mRNA 和 (pro) 肾素蛋白水平的增加。此外,佛波醇 12-肉豆蔻酸 13-乙酸酯激活蛋白激酶 C 可使肾素表达增加到与 Ang II 相同的程度。这些数据表明,Ang II 型 1 受体介导的 CD 肾素增加是由 Ang II 通过蛋白激酶 C 途径直接诱导的,这种调节不依赖于矿物皮质激素受体激活或上皮钠通道活性。