Department of Veterinary Pathobiology, College of Veterinary Medicine, Texas A&M University, College Station, Texas, United States of America.
PLoS One. 2011 Jan 19;6(1):e15811. doi: 10.1371/journal.pone.0015811.
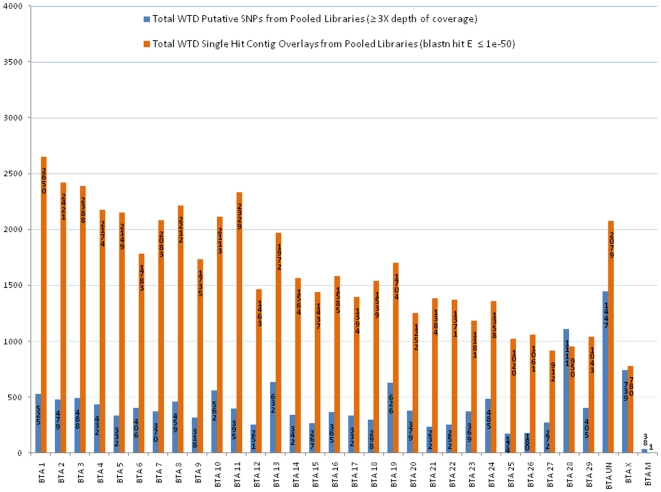
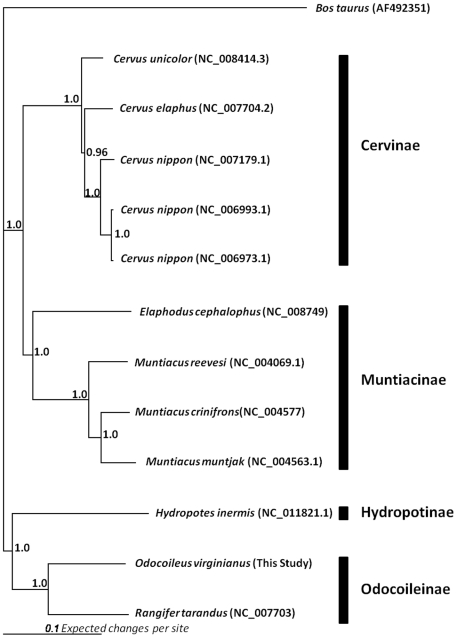
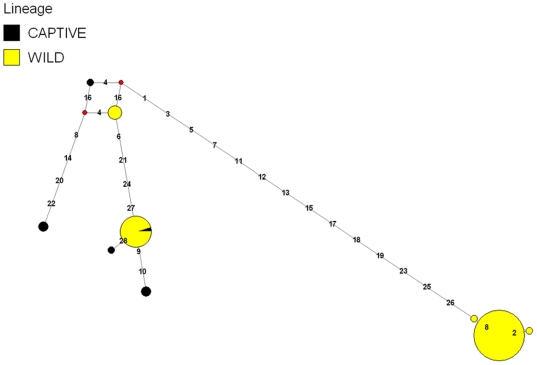
The white-tailed deer (Odocoileus virginianus) represents one of the most successful and widely distributed large mammal species within North America, yet very little nucleotide sequence information is available. We utilized massively parallel pyrosequencing of a reduced representation library (RRL) and a random shotgun library (RSL) to generate a complete mitochondrial genome sequence and identify a large number of putative single nucleotide polymorphisms (SNPs) distributed throughout the white-tailed deer nuclear and mitochondrial genomes. A SNP validation study designed to test specific classes of putative SNPs provides evidence for as many as 10,476 genome-wide SNPs in the current dataset. Based on cytogenetic evidence for homology between cow (Bos taurus) and white-tailed deer chromosomes, we demonstrate that a divergent genome may be used for estimating the relative distribution and density of de novo sequence contigs as well as putative SNPs for species without draft genome assemblies. Our approach demonstrates that bioinformatic tools developed for model or agriculturally important species may be leveraged to support next-generation research programs for species of biological, ecological and evolutionary importance. We also provide a functional annotation analysis for the de novo sequence contigs assembled from white-tailed deer pyrosequencing reads, a mitochondrial phylogeny involving 13,722 nucleotide positions for 10 unique species of Cervidae, and a median joining haplotype network as a putative representation of mitochondrial evolution in O. virginianus. The results of this study are expected to provide a detailed template enabling genome-wide sequence-based studies of threatened, endangered or conservationally important non-model organisms.
白尾鹿(Odocoileus virginianus)是北美分布最广、最成功的大型哺乳动物之一,但可用的核苷酸序列信息非常有限。我们利用简化代表性文库(RRL)和随机鸟枪法文库(RSL)的大规模平行焦磷酸测序,生成了完整的线粒体基因组序列,并鉴定出了大量分布在白尾鹿核和线粒体基因组中的假定单核苷酸多态性(SNP)。一项旨在测试特定类别假定 SNP 的 SNP 验证研究为当前数据集提供了多达 10,476 个全基因组 SNP 的证据。基于牛(Bos taurus)和白尾鹿染色体同源性的细胞遗传学证据,我们证明了一个分歧的基因组可用于估计从头序列连续图谱以及无草案基因组组装的假定 SNP 的相对分布和密度。我们的方法表明,为模式或农业重要物种开发的生物信息学工具可以用于支持具有生物学、生态学和进化重要性的物种的下一代研究计划。我们还提供了白尾鹿焦磷酸测序reads 组装的从头序列连续图谱的功能注释分析、涉及 13722 个核苷酸位置的 10 种独特鹿科物种的线粒体系统发育以及作为 O. virginianus 线粒体进化的假定代表的中位数连接单倍型网络。这项研究的结果有望提供一个详细的模板,用于对受到威胁、濒危或具有保护意义的非模式生物进行基于全基因组序列的研究。