Software College, Shenyang Normal University, Shenyang, PR China.
BMC Bioinformatics. 2011 Feb 2;12:44. doi: 10.1186/1471-2105-12-44.
Prediction of protein subcellular localization generally involves many complex factors, and using only one or two aspects of data information may not tell the true story. For this reason, some recent predictive models are deliberately designed to integrate multiple heterogeneous data sources for exploiting multi-aspect protein feature information. Gene ontology, hereinafter referred to as GO, uses a controlled vocabulary to depict biological molecules or gene products in terms of biological process, molecular function and cellular component. With the rapid expansion of annotated protein sequences, gene ontology has become a general protein feature that can be used to construct predictive models in computational biology. Existing models generally either concatenated the GO terms into a flat binary vector or applied majority-vote based ensemble learning for protein subcellular localization, both of which can not estimate the individual discriminative abilities of the three aspects of gene ontology.
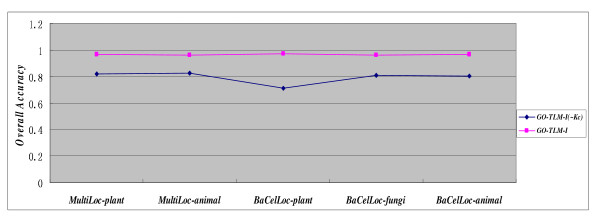
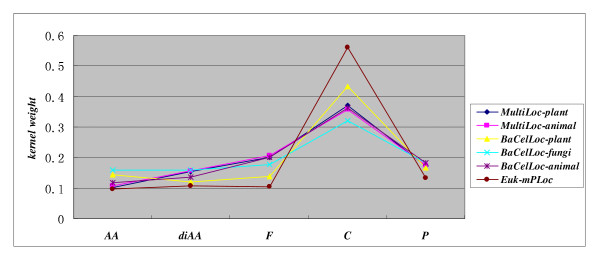
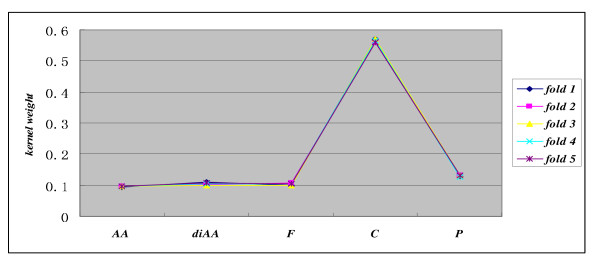
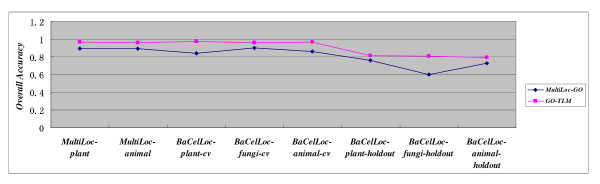
In this paper, we propose a Gene Ontology Based Transfer Learning Model (GO-TLM) for large-scale protein subcellular localization. The model transfers the signature-based homologous GO terms to the target proteins, and further constructs a reliable learning system to reduce the adverse affect of the potential false GO terms that are resulted from evolutionary divergence. We derive three GO kernels from the three aspects of gene ontology to measure the GO similarity of two proteins, and derive two other spectrum kernels to measure the similarity of two protein sequences. We use simple non-parametric cross validation to explicitly weigh the discriminative abilities of the five kernels, such that the time & space computational complexities are greatly reduced when compared to the complicated semi-definite programming and semi-indefinite linear programming. The five kernels are then linearly merged into one single kernel for protein subcellular localization. We evaluate GO-TLM performance against three baseline models: MultiLoc, MultiLoc-GO and Euk-mPLoc on the benchmark datasets the baseline models adopted. 5-fold cross validation experiments show that GO-TLM achieves substantial accuracy improvement against the baseline models: 80.38% against model Euk-mPLoc 67.40% with 12.98% substantial increase; 96.65% and 96.27% against model MultiLoc-GO 89.60% and 89.60%, with 7.05% and 6.67% accuracy increase on dataset MultiLoc plant and dataset MultiLoc animal, respectively; 97.14%, 95.90% and 96.85% against model MultiLoc-GO 83.70%, 90.10% and 85.70%, with accuracy increase 13.44%, 5.8% and 11.15% on dataset BaCelLoc plant, dataset BaCelLoc fungi and dataset BaCelLoc animal respectively. For BaCelLoc independent sets, GO-TLM achieves 81.25%, 80.45% and 79.46% on dataset BaCelLoc plant holdout, dataset BaCelLoc plant holdout and dataset BaCelLoc animal holdout, respectively, as compared against baseline model MultiLoc-GO 76%, 60.00% and 73.00%, with accuracy increase 5.25%, 20.45% and 6.46%, respectively.
Since direct homology-based GO term transfer may be prone to introducing noise and outliers to the target protein, we design an explicitly weighted kernel learning system (called Gene Ontology Based Transfer Learning Model, GO-TLM) to transfer to the target protein the known knowledge about related homologous proteins, which can reduce the risk of outliers and share knowledge between homologous proteins, and thus achieve better predictive performance for protein subcellular localization. Cross validation and independent test experimental results show that the homology-based GO term transfer and explicitly weighing the GO kernels substantially improve the prediction performance.
蛋白质亚细胞定位的预测通常涉及许多复杂因素,仅使用一个或两个方面的数据信息可能无法说明真实情况。出于这个原因,一些最近的预测模型被故意设计成集成多个异构数据源,以利用蛋白质特征信息的多方面。GO(基因本体)使用控制词汇表根据生物过程、分子功能和细胞成分来描述生物分子或基因产物。随着注释蛋白质序列的快速扩展,GO 已经成为一种通用的蛋白质特征,可以用于构建计算生物学中的预测模型。现有的模型通常要么将 GO 术语串联成一个平面二进制向量,要么应用基于多数投票的集成学习来进行蛋白质亚细胞定位,这两种方法都不能估计基因本体的三个方面的个体判别能力。
在本文中,我们提出了一种基于基因本体的转移学习模型(GO-TLM)用于大规模蛋白质亚细胞定位。该模型将基于特征的同源 GO 术语转移到目标蛋白质上,并进一步构建一个可靠的学习系统,以减少潜在的假 GO 术语的不利影响,这些术语是由于进化分歧而产生的。我们从基因本体的三个方面导出三个 GO 核,以测量两个蛋白质之间的 GO 相似性,并导出另外两个谱核,以测量两个蛋白质序列之间的相似性。我们使用简单的非参数交叉验证来显式地权衡五个核的判别能力,从而大大降低了与复杂半定规划和半不定线性规划相比的时间和空间计算复杂度。然后,将这五个核线性合并为一个用于蛋白质亚细胞定位的单个核。我们在基准数据集上评估了 GO-TLM 与三个基线模型(MultiLoc、MultiLoc-GO 和 Euk-mPLoc)的性能。5 折交叉验证实验表明,GO-TLM 与基线模型相比取得了显著的准确性提高:在数据集 MultiLoc 植物和数据集 MultiLoc 动物上,分别比模型 Euk-mPLoc 提高了 80.38%和 12.98%;在数据集 MultiLoc-GO 上分别提高了 96.65%和 96.27%,比模型 MultiLoc 提高了 7.05%和 6.67%;在数据集 BaCelLoc 植物、数据集 BaCelLoc 真菌和数据集 BaCelLoc 动物上,分别比模型 MultiLoc-GO 提高了 13.44%、5.8%和 11.15%。对于 BaCelLoc 独立集,GO-TLM 在数据集 BaCelLoc 植物保留集、数据集 BaCelLoc 植物保留集和数据集 BaCelLoc 动物保留集上的准确率分别为 81.25%、80.45%和 79.46%,而基线模型 MultiLoc-GO 的准确率分别为 76%、60.00%和 73.00%,准确率分别提高了 5.25%、20.45%和 6.46%。
由于直接基于同源的 GO 术语转移可能容易将噪声和异常值引入目标蛋白质,因此我们设计了一个显式加权核学习系统(称为基于基因本体的转移学习模型,GO-TLM),将相关同源蛋白质的已知知识转移到目标蛋白质上,这可以降低异常值的风险,并在同源蛋白质之间共享知识,从而实现蛋白质亚细胞定位的更好预测性能。交叉验证和独立测试实验结果表明,基于同源的 GO 术语转移和显式加权 GO 核显著提高了预测性能。