Software College, Shenyang Normal University, Shenyang, China.
PLoS One. 2013 Nov 18;8(11):e79606. doi: 10.1371/journal.pone.0079606. eCollection 2013.
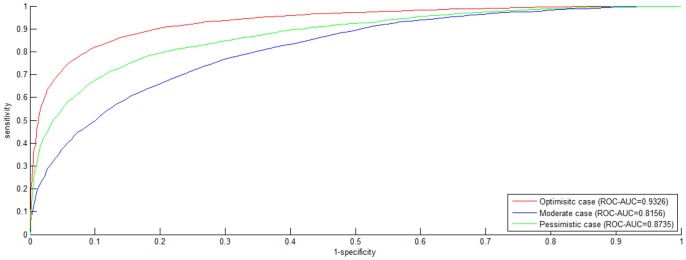
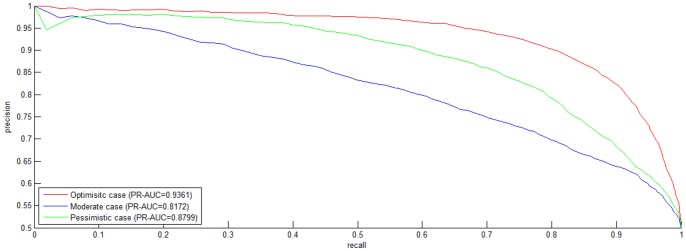
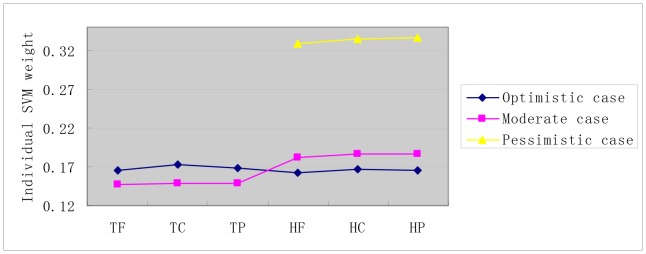
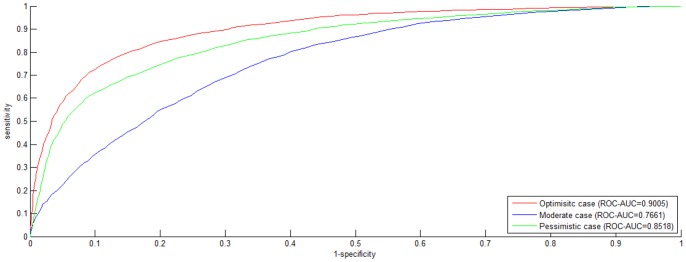
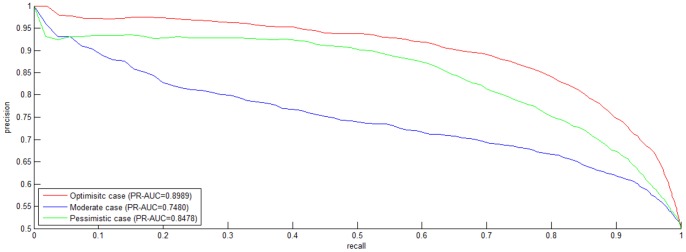
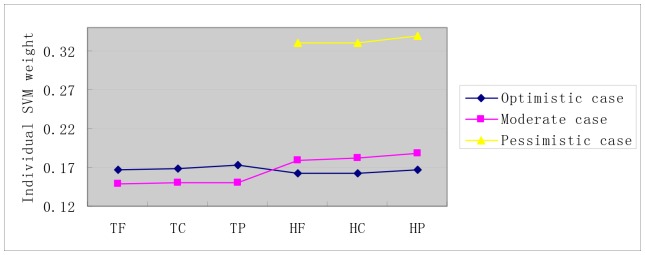
Reconstruction of host-pathogen protein interaction networks is of great significance to reveal the underlying microbic pathogenesis. However, the current experimentally-derived networks are generally small and should be augmented by computational methods for less-biased biological inference. From the point of view of computational modelling, data scarcity, data unavailability and negative data sampling are the three major problems for host-pathogen protein interaction networks reconstruction. In this work, we are motivated to address the three concerns and propose a probability weighted ensemble transfer learning model for HIV-human protein interaction prediction (PWEN-TLM), where support vector machine (SVM) is adopted as the individual classifier of the ensemble model. In the model, data scarcity and data unavailability are tackled by homolog knowledge transfer. The importance of homolog knowledge is measured by the ROC-AUC metric of the individual classifiers, whose outputs are probability weighted to yield the final decision. In addition, we further validate the assumption that only the homolog knowledge is sufficient to train a satisfactory model for host-pathogen protein interaction prediction. Thus the model is more robust against data unavailability with less demanding data constraint. As regards with negative data construction, experiments show that exclusiveness of subcellular co-localized proteins is unbiased and more reliable than random sampling. Last, we conduct analysis of overlapped predictions between our model and the existing models, and apply the model to novel host-pathogen PPIs recognition for further biological research.
宿主-病原体蛋白相互作用网络的重建对于揭示潜在的微生物发病机制具有重要意义。然而,目前实验得出的网络通常较小,应该通过计算方法来进行扩充,以进行更无偏的生物学推断。从计算模型的角度来看,宿主-病原体蛋白相互作用网络的重建存在数据稀缺、数据不可用和负样本数据采样三个主要问题。在这项工作中,我们旨在解决这三个问题,并提出了一种用于 HIV-人类蛋白相互作用预测的概率加权集成转移学习模型(PWEN-TLM),其中支持向量机(SVM)被用作集成模型的个体分类器。在该模型中,通过同源知识转移来解决数据稀缺和数据不可用的问题。同源知识的重要性通过个体分类器的 ROC-AUC 度量来衡量,其输出被概率加权以得出最终决策。此外,我们进一步验证了这样的假设,即只有同源知识就足以训练出用于宿主-病原体蛋白相互作用预测的令人满意的模型。因此,该模型对数据不可用具有更强的鲁棒性,对数据约束的要求也较低。关于负样本数据的构建,实验表明,亚细胞共定位蛋白的排他性是无偏的,比随机采样更可靠。最后,我们分析了我们的模型与现有模型之间的重叠预测,并将该模型应用于新的宿主-病原体 PPIs 识别,以进行进一步的生物学研究。